Isolation
/ Synthesis / Detection of Nucleic Acids
DNA
the old fashioned way - Genomic DNA from Bacteria
Many biochemistry and molecular biology procedures are now available on the web. Here is one such
reference web site at NWFSC. Isolation of DNA takes advantage of its different size and solubility properties
compared to lipids, proteins and smaller biomolecules.
grow cells / centrifuge / lyze cells (add SDS)
separate proteins and polysaccharides - add CTAB (cetyl tirmethyl ammonium bromide)
partition using chloroform / isoamyl alcohol
add ethanol or isopropanol / ppt nucleic acid / isolate DNA
Synthesis of DNA of Specified Sequence
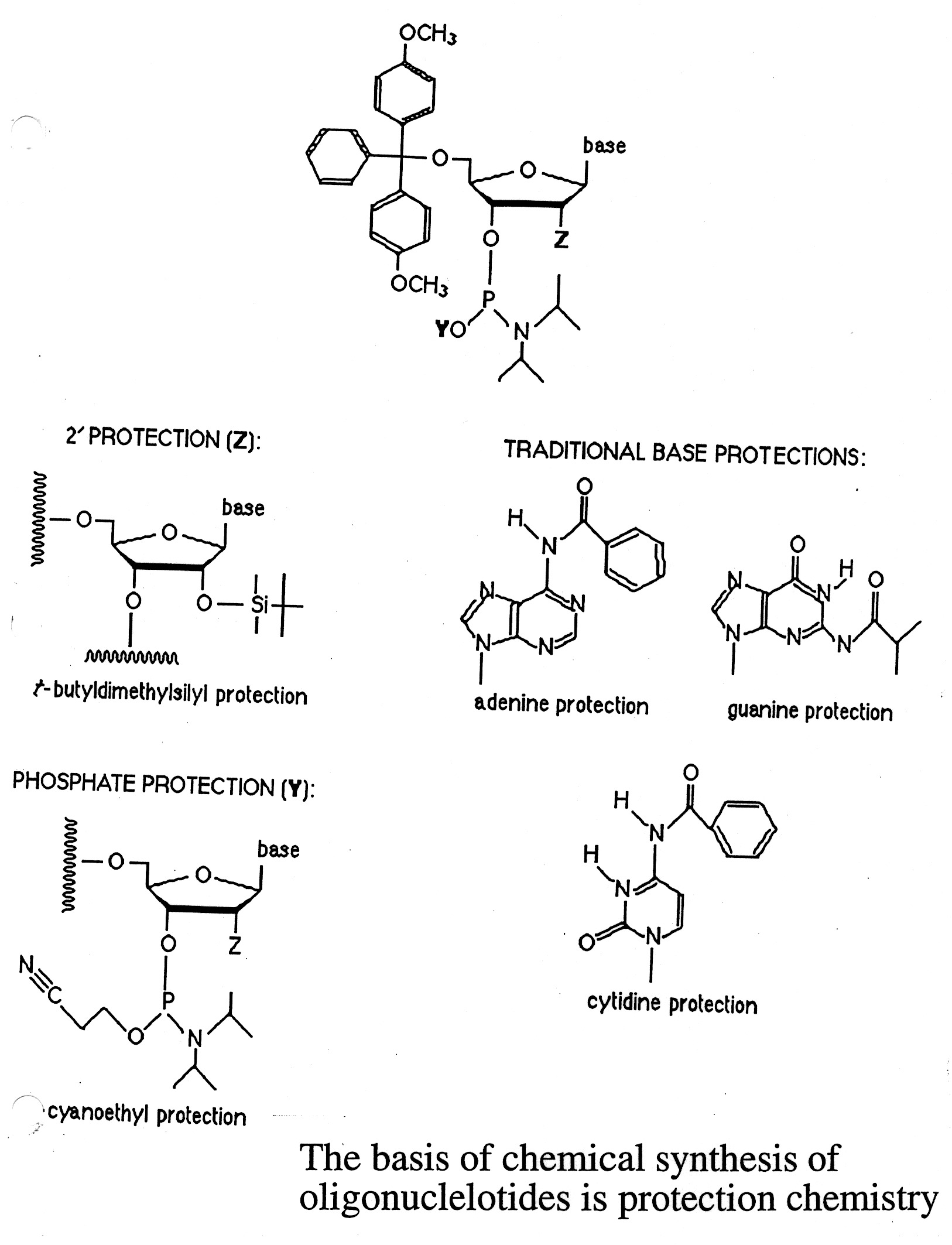
The chemical synthesis of nucleic acids is based on the use of phosphoramidites. The synthesis of DNA is done in
iterative cycles on a solid support. The key to chemical synthesis is the "blocking" and "unblocking" of reactive
groups through the cycle - protective chemistry:
- C5' hydroxyl / DMT (dimethoxy trityl)
- N bases / benzoyl (dA and dC) or isobutyryl (dG)
- Phosphite group / cyano ethyl
The chemical synthesis proceeds from the 3' to 5' direction in an iterative process (detritylation / activation /
coupling / capping / and oxidation steps) (see U. Penn. web site link for details).
Synthesis consists of a cycle of 4-steps ( see below):
"deblocking" to remove DMT
"activation" / "coupling"
"capping" - using acetic anhydride (if growing chain does not elongate, it gets capped and eliminated)
"oxidation" - phosphate
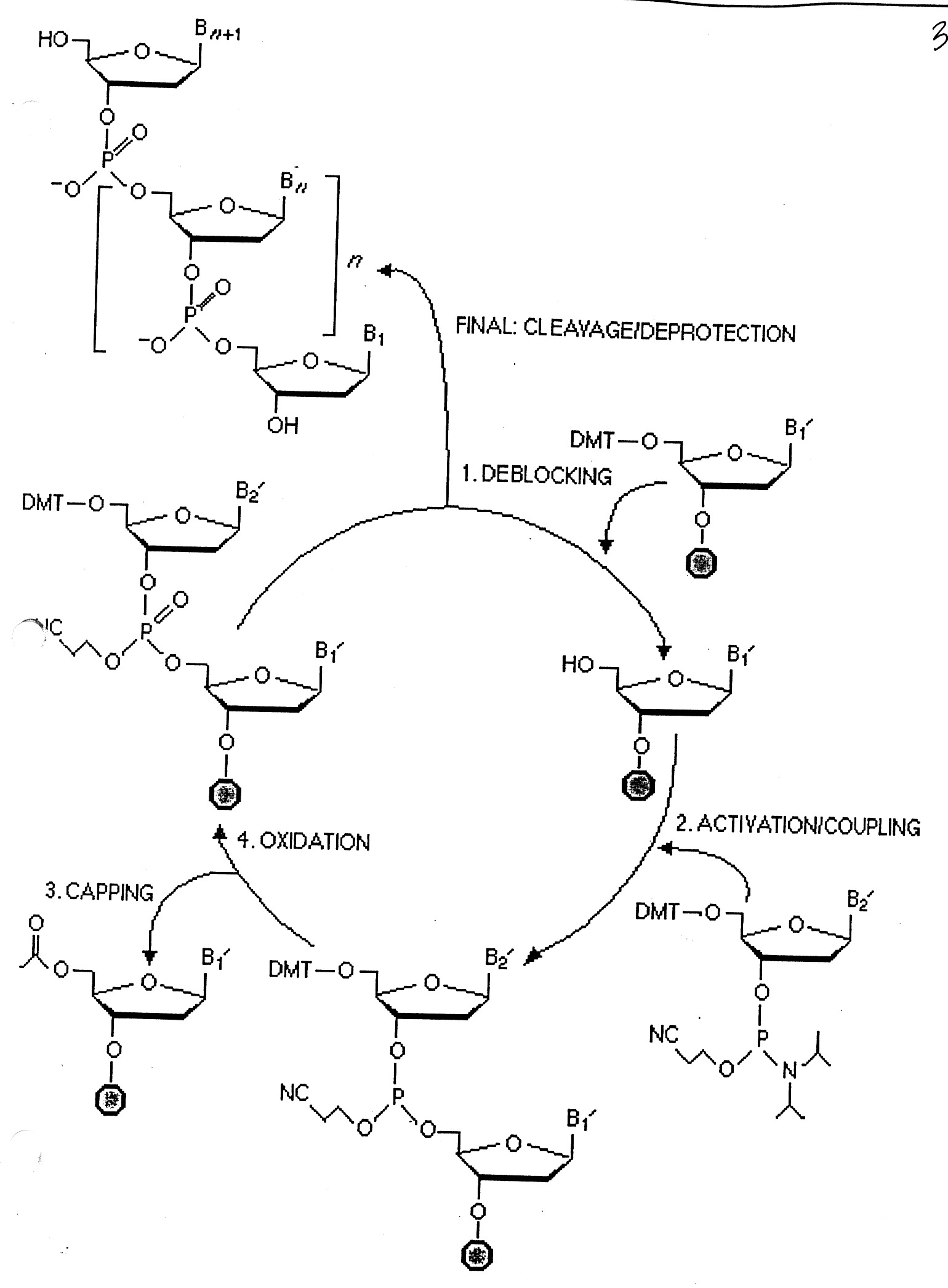
At the end of the synthesis, the new oligo is released from the solid support and deblocked ((NH4OH at 55o for 8 hours).
The "Yield" is dependent on the efficiency of each step for multiple cycles.
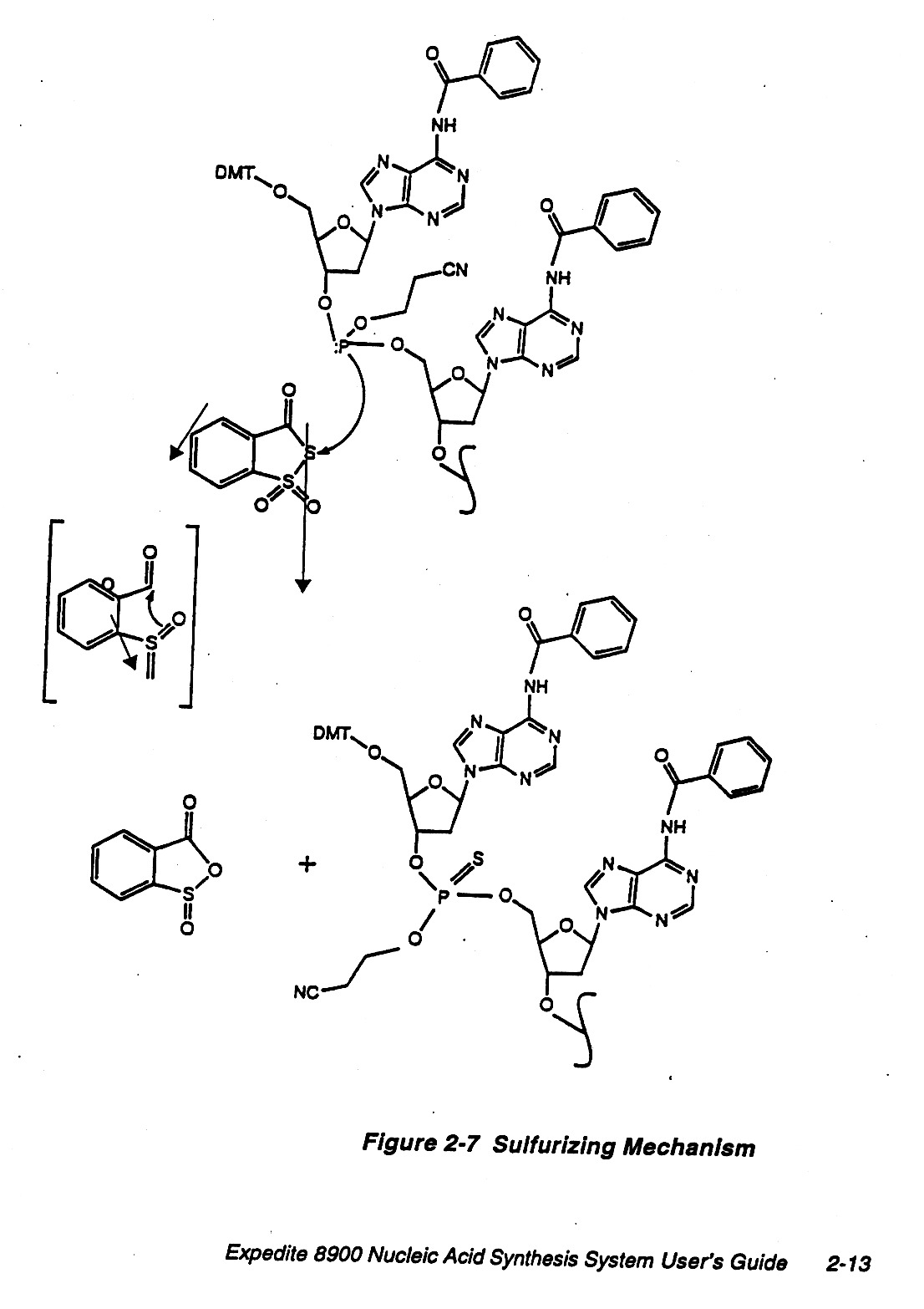
Chemically DNA is fairly stable but in our cells there are nucleases, etc. that are used to recycle nucleotides
and also for protection. In the laboratory one can employ other linkages that are more resistant to
nucleases, e.g. sulfurizing method.
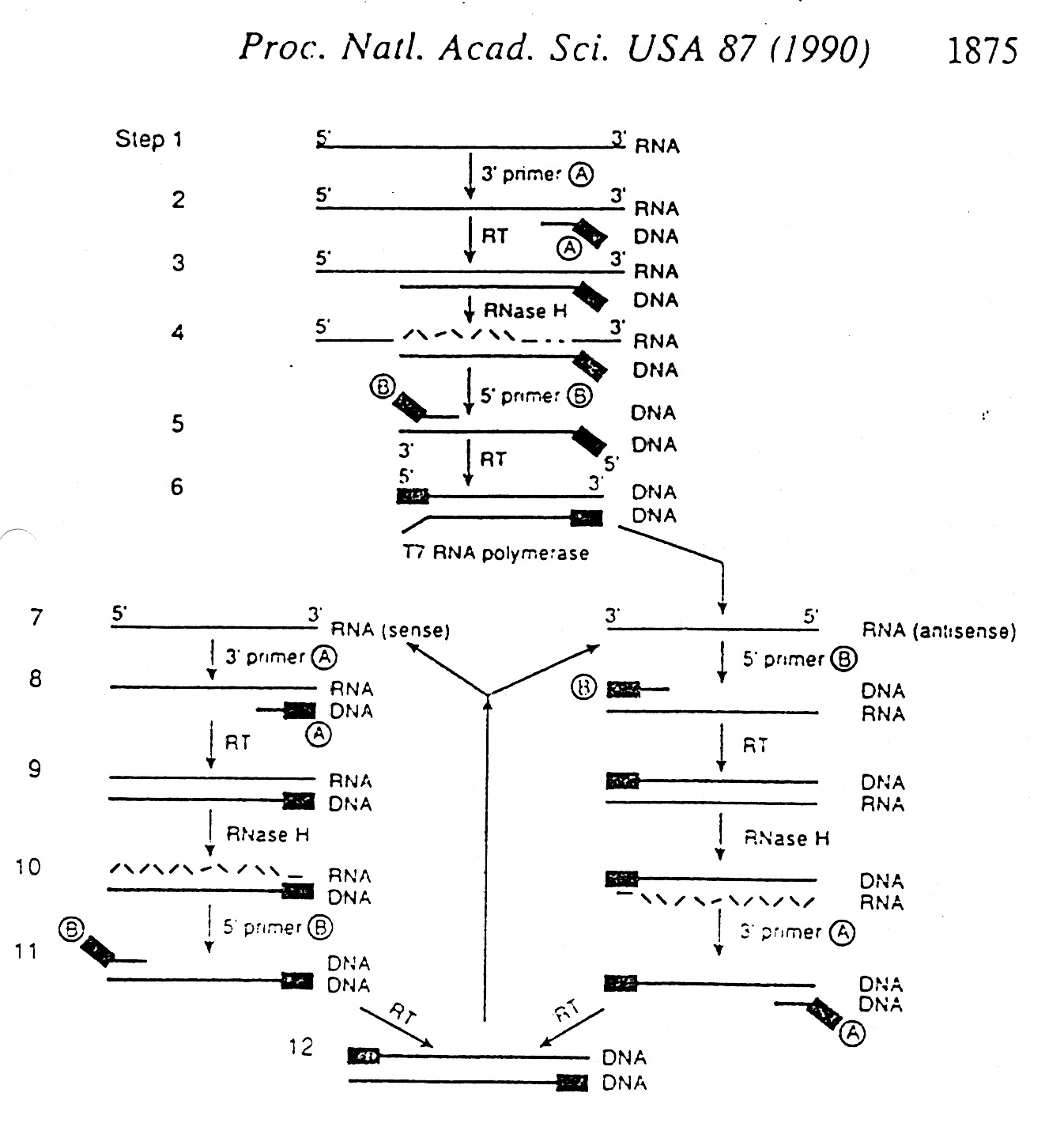
Amplification of DNA
Several Methods: Cloning
PCR (Polymerase Chain Reaction)
Other amplification methods take advantage of RT (reverse transcriptase) and
T7 RNA Polymerase (makes many copies of RNA per cycle).
Analysis of DNA - use of radio isotopes
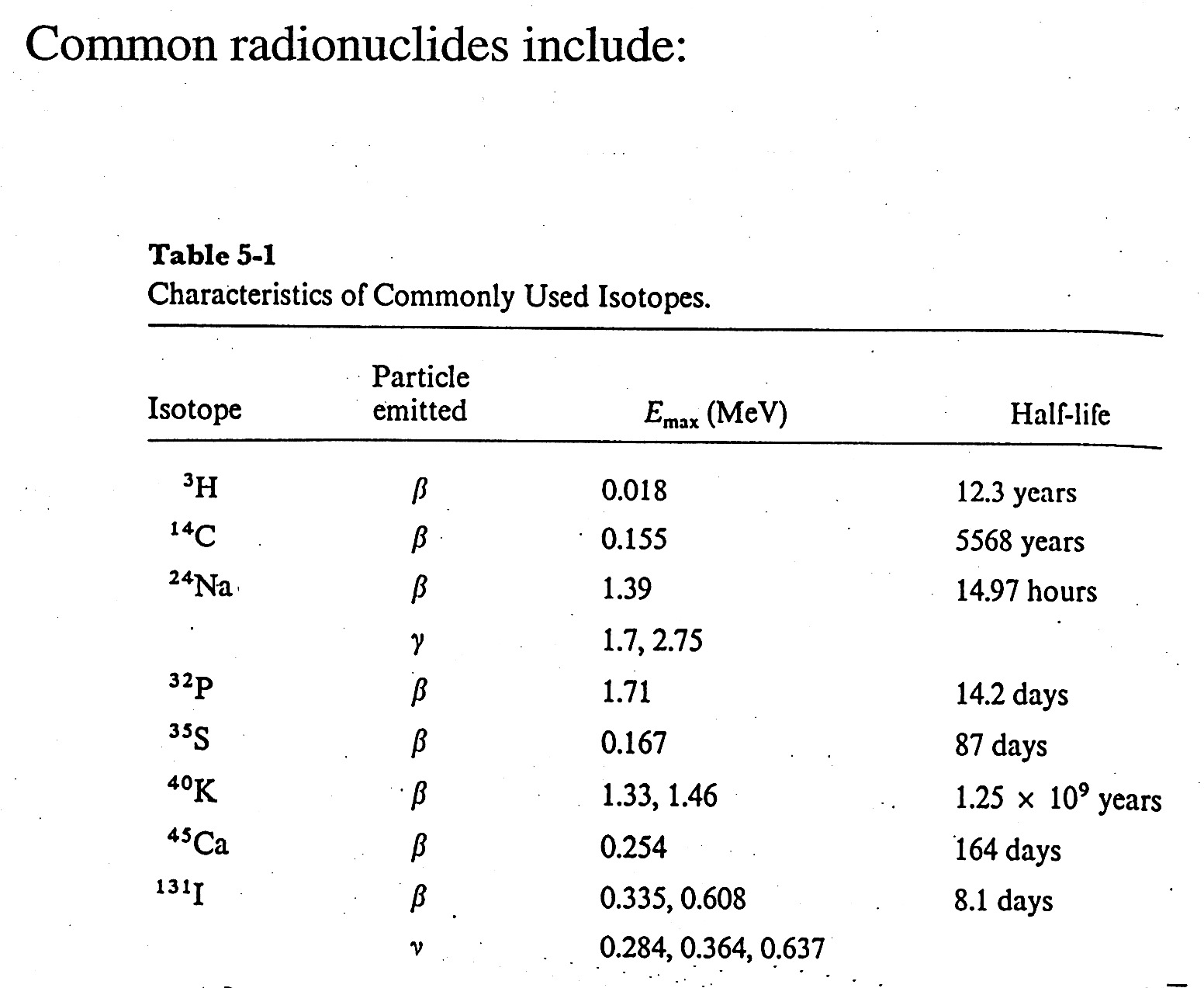
How to Define Units for Radioactivity
- each isotope has a characteristic decay / characteristic decay particle, E, and half-life.
- for nucleic acid chemistry - 32P is the most common and useful isotope;
beta emitter / 1.71 MeV, half-life = 14.2 days
- Radioactivity is measured in Curies (Ci); 1 Ci = 3.7 x 1010 disintegrations / second
1 microCurie ( 1mCi = 2.22 x 106 disintegrations / minute
Define Specific Activity = disintegrations / unit mass = mCi / 1mmole
In practice, instruments measure counts per minute (CPM) which is related to DPM.
How to Measure Radioactivity
1. Geiger Counter: count ions produced to in a gas chamber by the passage of a decay particle
2. Scintillation Counter: Embed radioactive sample totally within chamber, usually in a solvent (toluene / p-Xylene / 1,4-Dioxane) mixed with a "fluor" (PPO / POPOP). The radioactive decay particle excites many solvent molecules as it looses energy, these in turn excite the fluors (POP / POPOP) used in scintillation counting which emit fluoresence at wavelengths that can be readily counted by surrounding photomultipliers. Reduce background from thermal noise by using "coincidence counters". Another advantage of liquid scintillation counters is that the number of photons produced is roughly proportional to the energy of the beta particle counted, so the counter can be "set" to detect different kinds of decay by setting "energy windows" on the detector.
3. Film: Overlay photographic film over radioactive sample, the intensity ("blackening") of the film gives a rough measure of the amount of radioactivity in the sample.
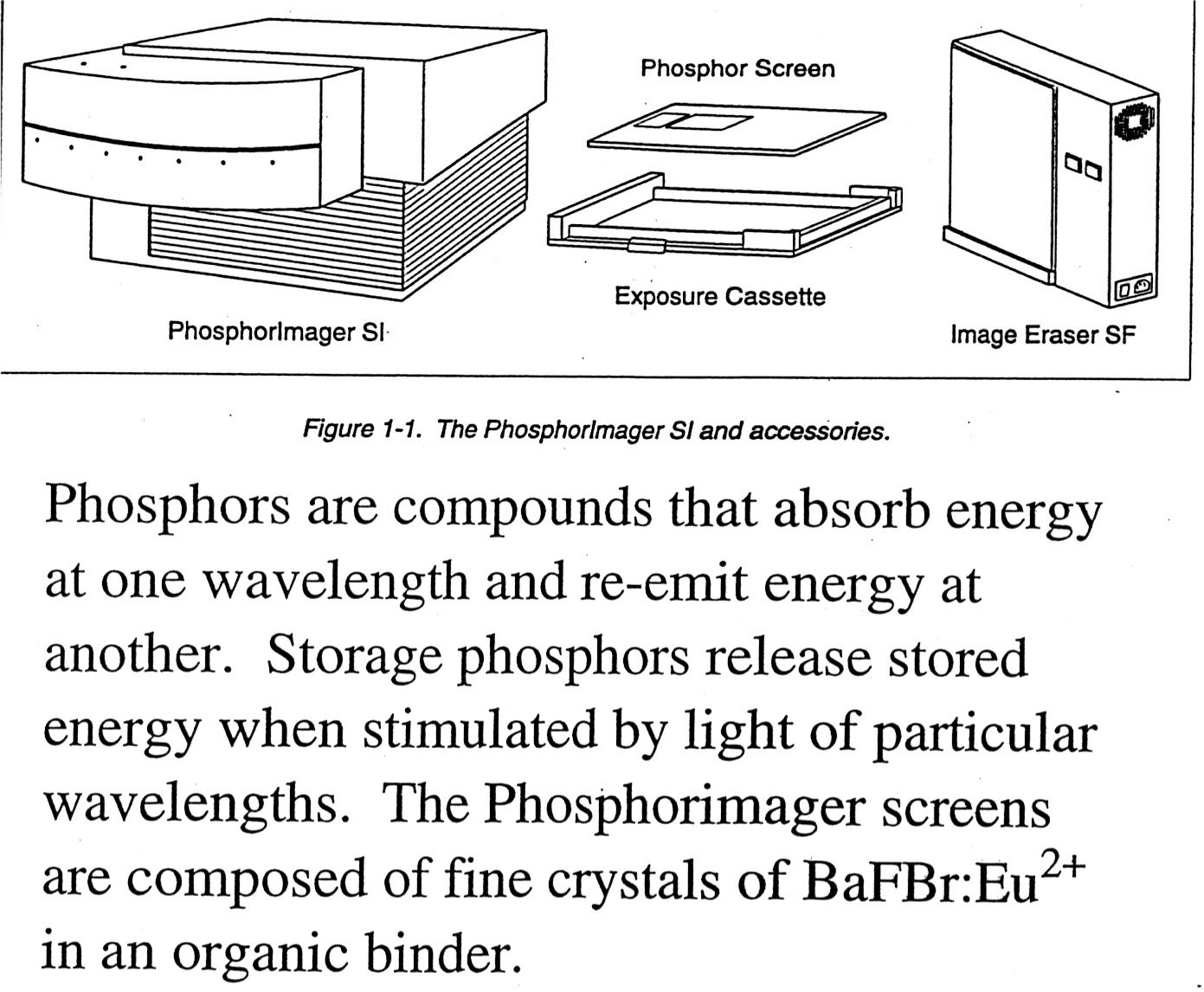
4. PhosphorImagers: Use a PhosphorImager Screen to take the place of film. Advantages are that the PI screen is more sensitive than film, can be exposed in less time, and is reusable. The phosphors on the screen absorb energy at one wave-length and emit it at another that can be read out by photomultipliers. Phosphorimager screens are composed of fine crystals of BaRBr:Eu+2. Radioactive sample excites the Eu+2 and mobile electrons become trapped in "storage" phosphors that can retain the "trapped" signal for up to a couple of hours. An instrument called the "Phosphorimager" scans the screen using a helium-neon laser that emits red light at 633 nm. The charged BaFBr- complexes absorb the red light, the trapped electrons are freed, Eu+3 is reduced back to Eu+2* and as Eu+2* returns to the ground state Eu+2 it releases energy as blue light in proportion to the initial radioactivity that is then collected by the Photomultiplier Tube (PMT). The scanned image and resulting PMT counts generate a "digital" image of the "film". The PI screen can be regenerated by exposure to strong light and reused almost indefinitely. (Molecular Dynamics is one maker of PhosphorImagers, find more details on the use of PI on their web site.).
How to Label Nucleic Acids
Nucleic acids are normally labeled using 32P by incorporating either alpha or gamma labeled nucleoside triphosphates.
Manipulation of DNA and RNA:
1. DNA Polymerase: Nick DNA with DNase, remove nucleotides by 5'-->3' exonuclease activity of E. coli DNA polymerase I. Replace excised nucleotides with alpha radiolabeled nucleotides by E. coli DNA polymerase I.
2. Use of Phosphatases and Kinases: Use gamma labeled nucleotide to label 5' phosphate.
Separation of Nucleic Acids - use of Gel Electrophoresis
Electrophoresis refers to moving charged particles due to an applied voltage. The force experienced by the particle is given by Coulomb's Law:
F = z e E (z = # charges; e = electronic charge; E = electric potential)
at steady state velocity, Force of acceleration due to potential = Force of friction = f(vel) where f is the frictional coefficient, so f(vel) = zeE. The electrophoretic mobility (m) is the ratio of the velocity (vel.) of the particle to the electrical potential (E).
(m) = vel/E = ze/f
Frictional coefficients can be calculated for a variety of sizes, but in practice biochemists rely on relative mobilities - that is the mobility relative to that of reference standards of known molecular weights. A plot of relative mobility vs. log(MW) is nearly linear for restriction fragments of DNA.
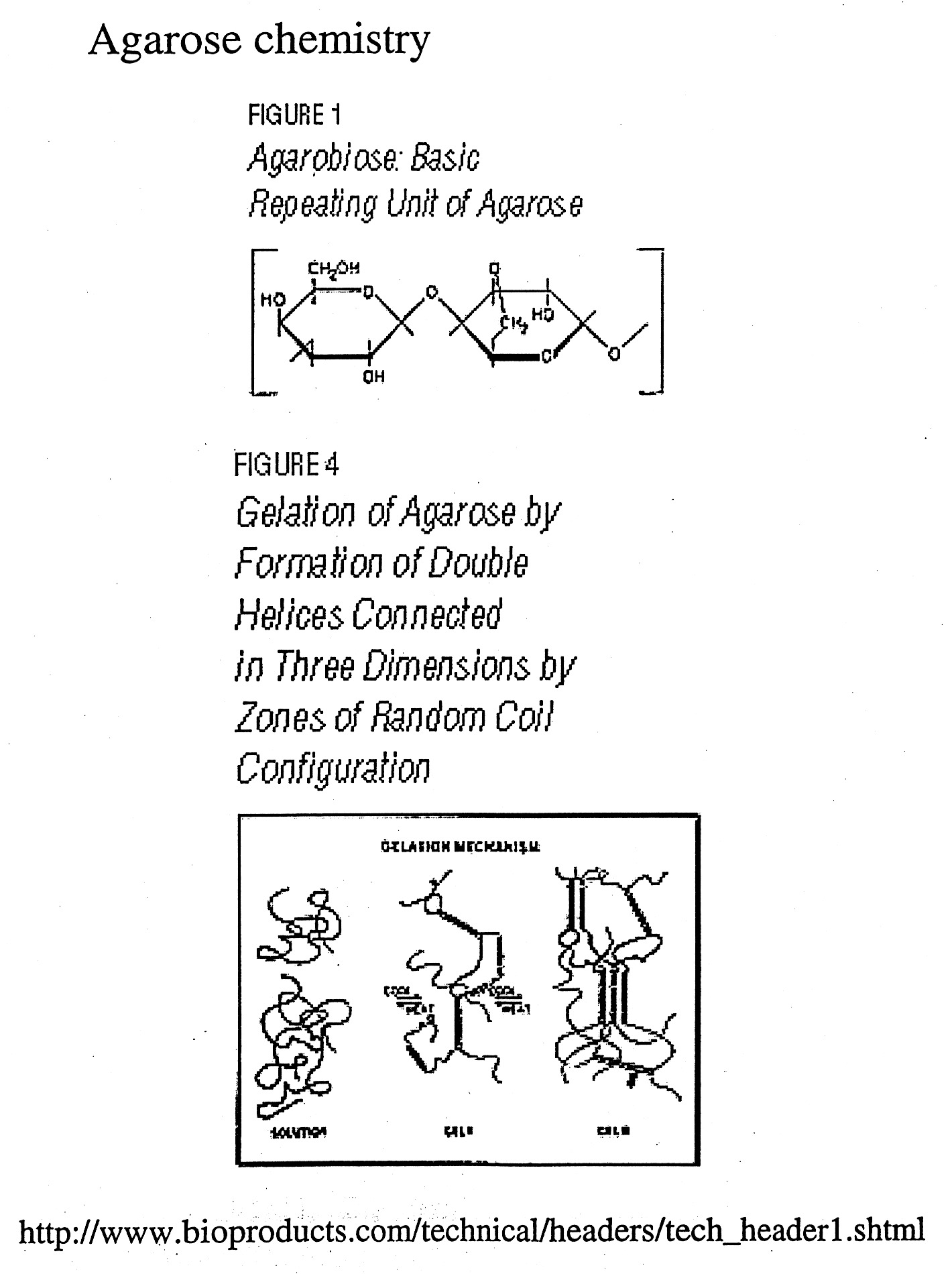
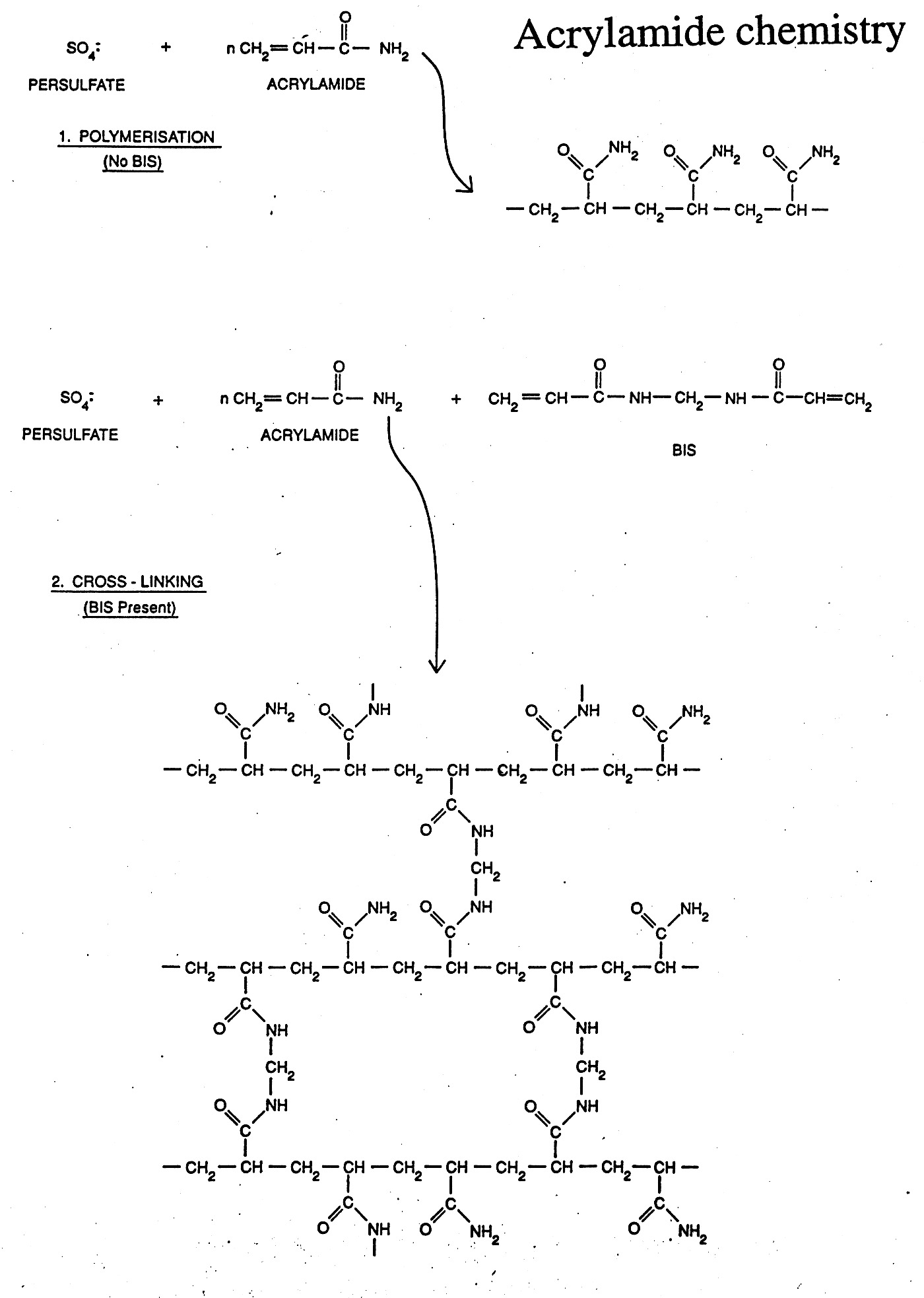
Gels can be made of a variety of materials (agarose, polyacrylamide, etc.) and cast in different ways (tube gels, vertical slab gels, horizontal slab gels, etc.) The porousity can be varied by varying the concentration (1% vs. 2% agarose) or % concentration and % cross-linking as in the case of polyacrylamide gels.
Gel electrophoresis uses the porousity of the gel to separate particles by size (actually their charge / size (mass) ratios). Since nucleic acids carry one negative charge per nucleotide and thus have nearly uniform charge/mass ratios, nucleic acids can be readily separated in the gel by size (length) if the shapes are uniform.
Gel Electrophoresis can be carried out under "Native" or "Denaturing" (SDS / Urea) conditions. During native gel electrophoresis samples retain native or near native conformations and charges, thus mobility is usually proportional for double-stranded DNA, but highly unpredictable for native proteins and single-stranded nucleic acids which can fold back on themselves to form a variety of secondary structures. However, nucleic acids run under denaturing conditions (7M urea) separate uniformly according to length. SDS is used to denature proteins and coat the protein with negative charge, mimicking the situation naturally occurring with nucleic acids. This generates nearly uniform charge to mass ratios for proteins and thus allows them to be separated by size (more on this later).
************************