 -phenylalanine
-phenylalanineIf a meteorite contains optically active compounds this would suggest life elsewhere in the universe.
-phenylalanine
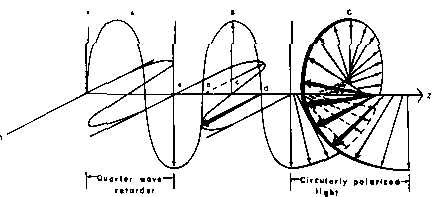
Passing plane polarized light through a birefringent plate (in the z-direction) which splits the light into two plane-polarized beams oscillating along different axes (e.g., fast along x and slow along y). When one of the beams is retarded by 90º (using a quarter-wave retarder) then the two beams which are now 90º out of phase are added together, the result is circularly polarized light of one direction. By inverting the two axes such that the alternate beam is retarded than circularly polarized light of the other direction is generated.
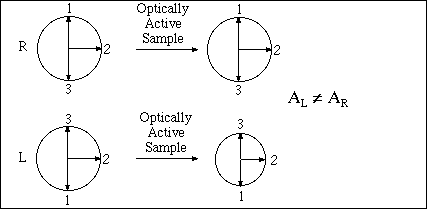
The result of adding the right and left circularly polarized that passes through the optically active sample is elliptically polarized light, thus circular dichroism is equivalent to ellipticity.
It is now possible to generate a wide variety of different proteins via solid-phase synthesis and recombinant DNA methods. There is a need to determine the structure of larger and larger numbers of proteins. THigh resolution techniques such as NMR and X-ray crystallography (that are complex and time-consuming) will be overwhelmed. Therefore, there is a need for quick low resolution techniques for determining protein structure. For example, absorption, Raman, fluorescence, and circular dichroism (CD).
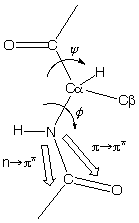
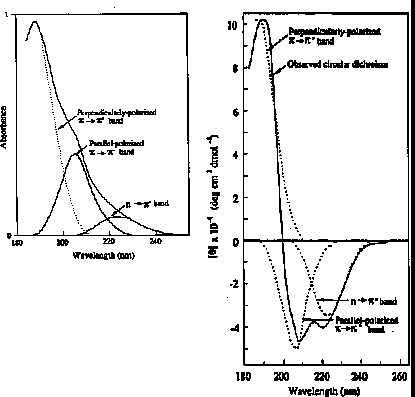
n -> p* centered around 220 nm
p -> p* centered around 190 nm
n -> p* involves non-bonding electrons of O of the carbonyl
p -> p* involves the p-electrons of the carbonyl
The intensity and energy of these transitions depends on f and y (i.e., secondary structure)
For example, the absorption spectrum of poly-L-lysine in an a-helix, b-sheet, and unordered (random coil) differ due to long-range order in the amide chromophore.
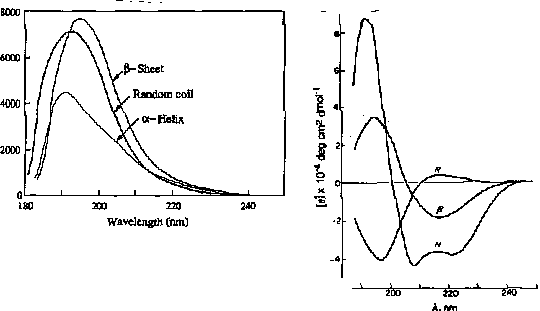
positive at 212 nm (p->p*)
negative at 195 nm (n->p*)
negative at 218 nm (p->p*)
positive at 196 nm (n->p*)
exiton coupling of the p->p* transitions leads to positive (p->p*)perpendicular at 192 nm and negative (p->p*)parallel at 209 nm
negative at 222 nm is red shifted (n->p*)
generate basis sets by determining spectra of pure a-helix, b-sheet, etc. of synthetic peptides
or deconvoluting CD spectra of proteins with know structures to generate basis sets of each of secondary structure
random coil at pH 7.0 / a-helix at pH 10.8 / b-form at pH 11.1 after heating to 52°C and recooling
Recently Sa(l) was derived from the spectrum of myoglobin which is 80% a-helix
Recently b-turn has been added to the above equation {ST(l)}. ST(l) was derived from a combination of L-Pro-D-Ala, (Ala2-Gly2)n and Pro-Gly-Leu
b-form is now (Lys-Leu)n in 0.1 M NaF at pH 7
Random-coil is now (Pro-Lys-Leu-Lys-Leu)n in salt free neutral solution.
a correction of chain-length for a-helix has been recently introduced:
![]()
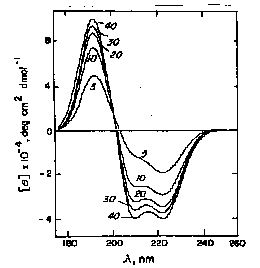
The disadvantage of this method is that although these basis sets are easily determined by direct measurement, they do not always agree from one lab to another. In addition, chain length and aggregation effect the basis set spectra. However, this method is usually accurate to within 10% for a-helix content.
Technique Secondary Structure carboxypeptidase a-chymotrypsin myoglobin a 23% 8% 68% X-ray b 18% 22% 0% RC + other 59% 70% 32% a 13% 12% 68% CD using (Lys)n Basis Sets b 31% 23% 5% RC + other 56% 65% 27%
spectrum of protein with known structure =
usually 5-15 proteins are used to generate basis set spectra
choice of reference proteins is arbitrary and effects results
determination of secondary structure from X-ray data is subject to error and disagreements among groups
secondary structures are NOT ideal in real proteins (e.g., a-helices can be bent, the spectrum of a 310-helix is different from a-helix, and b-turn can be twisted, non-planar, or perpendicular or parallel b-sheets.
If the aromatic residue is held rigidly in space than its environment is asymmetric, and it will exhibit circular dichroism.In a molten globule the far-UV CD is retained while the near-UV CD is lost.
Aromatics have allowed p->p* transitions (1La and 1Lb) that are directed in the plane of the p-bonding system and are orthogonal to each other.
Phe has a small extinction coefficient because of high symmetry and it is also the least sensitive to alterations in its environment. Absorption maxima at 254, 256, 262 and 267 nm (vibronic bands).
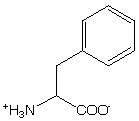
Tyr has lower symmetry then Phe and therefore has more intense absorption band. Tyr has absorption maximum at 276 nm and a shoulder at 283 nm. Hydrogen-bonding to the hydroxyl group leads to a red-shift of up to 4 nm. The dielectric constant effects the spectrum also.
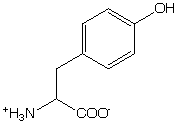
Trp has the most intense absorption band centered at 282 nm. Hydrogen-bonding to the NH can shift the 1La band by as much as 12 nm.
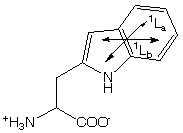
Disulfide (S-S) spectra have a broad band at 250 - 300 nm with no vibronic structure.
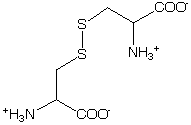
These groups can complicate the Far-UV CD region because of allowed p->p* transitions
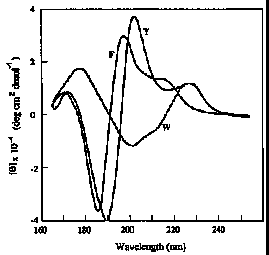
Vacuum ultraviolet CD spectra of the models of aromatic side chain residues. Glutamyl tyrosine (Y); lysyl-phenylalanine (F); glutamyl-tryptophan (W).
Summary
CD has an important role in the structural determinants of proteins
However, the effort expended in determining secondary structure elements is usually not worth it because it is somewhat unreliable.
The real power of CD is in the analysis of structural changes in a protein upon some perturbation, or in comparison of the structure of an engineered protein to the parent protein.
CD is rapid and can be used to analyze a number of candidate proteins from which interesting candidates can be selected for more detailed structural analysis like NMR or X-ray crystallography.
Protein secondary structure and circular dichroism: A practical guide
q = ellipticity
[q] = ellipticity =
, c = concentration, l = path length
q = 2.303 (AL - AR) 180/4p (in units of degrees)
[q]MRW =
(deg cm2 dmole-1) = mean residue weight ellipticity
n = number of amino acids
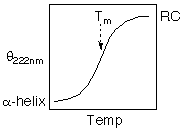
The thermodynamic parameters, DGu, DHu, DSu, Tm, DCp can be determined
DG = ((DH(1-(T/Tm)) - DCp((Tm-T) + T ln(T/Tm))
Fraction Unfolded = 1 - (1/(1 + (exp((DH + DCp (T-Tm) - T DH/Tm - T DCp ln(T/Tm))/R/-1/T))))
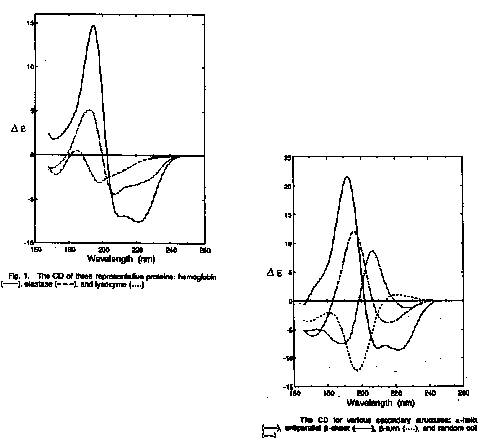
The goal is to determine the fraction of basis set spectra that add up to give the CD spectrum of the protein (hemoglobin, elastase, lysozyme)
The absorption should be less than 1.0 (usually < 0.3) for cell pathlengths of 0.05 to 1 cm in order to maintain reasonable signal-to-noise ratios and accurate CD measurements.
Protein concentration is typically 1 mg/mL
Buffer is typically 10 mM phosphate with low salt if any.
Need to know protein concentration accurately.
e190nm = 8,500 - 11,400 M-1 cm-1 per residue (this is not accurate enough: as you know the e in the far UV of proteins depends on the secondary structure).
e280nm (in 6 M GdmCl) = # of Trp residues ¥ 5,690 + # of Tyr residues ¥ 1,280 M-1 cm-1
| Protein | Technique | a-helix
H |
antiparallel-b-sheet
A |
parallel-b-sheet
P |
b-turn
T |
other
O |
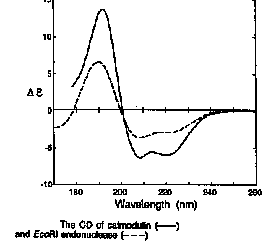
| EcoRI endonuclease | X-ray | 26 | 20 | 8 | 25 | 21 |
| EcoRI endonuclease | Deconvolution of CD spectrum | 33 | 20 | 5 | 17 | 25 |
| calmodulin | X-ray | 59 | 3 | 0 | - | 41 |
| calmodulin | Deconvolution of CD spectrum | 61 | 2 | 2 | - | 35 |
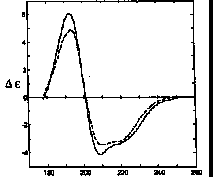
Thymidylate synthetase is 33% H, 24% A, 2% P, 21% T, 20% O. Upon binding of FdUMP and 5,10-methylenetetrahydrofolate CD shows -5% A, -6% T, +8% O.
The bases themselves are directly involved in close interactions in all common secondary structures.
Even some actual sequence information must be taken into account to explain CD spectra. For example the CD spectrum of the dinucleoside phosphate ApG is different from the sum of the CD spectra of A and G monomers.
The CD spectra of ApG and GpU are the sum of the two monomers plus and an additional term to account for the base-base interactions:
2[qApG(l)] = [qA(l)] + [qG(l)] + IAG(l)
2[qGpU(l)] = [qG(l)] + [qU(l)] + IGU(l)
In a trinucleoside diphosphate such as ApGpU, there would be contributions from three monomers, two interactions between neighboring bases, and a final contribution due to interaction between the next-nearest neighbors, A and U:
3[qApGpU(l)] = [qA(l)] + [qG(l)] + [qU(l)] + I'AG(l) + I'GU(l) + IAU(l)
One can assume that IAG(l) = I'AG(l) and that IAU(l) is negligible, therefore:
3[qApGpU(l)] = 2[qApG(l)] + 2[qGpU(l)] - [qG(l)]
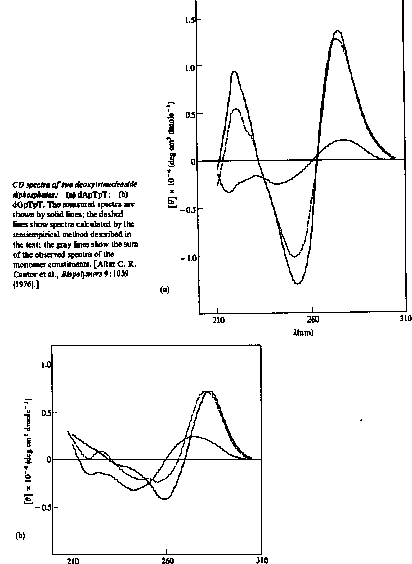
The spectrum of a single-strand stacked helix would contain optical contributions from each of the 16 possible dinucleoside phosphates, weighted by their frequencies of occurrence. The spectrum of a double-strand is accounted for in an analogous way, by adding the contributions of each of the 10 possible double-strand dimers (ApG base paired with CpU, and so on).
In practice, a total of up to 30 different spectral contributions must be combined to compute the CD of a molecule such as tRNA that has both single-strand and double-strand regions.
This approach is very complex!