Protein Folding and Molecular Interactions
Goals for this unit:
1.
Types of non-covalent Interactions
2.
Protein Folding - thermo
and approaches to predicting protein folds
3. Use of energy potentials and simulations
4. Denaturation / Renaturation - thermo and practice
5.
Role
of chaperones / models used to describe folding (molten globule)
Four Levels of Description of (Native) Protein Structure
Primary Structure: (~60-1000 amino acid residues)
linear
seq. of amino acid residues, covalent bonding including -SS-
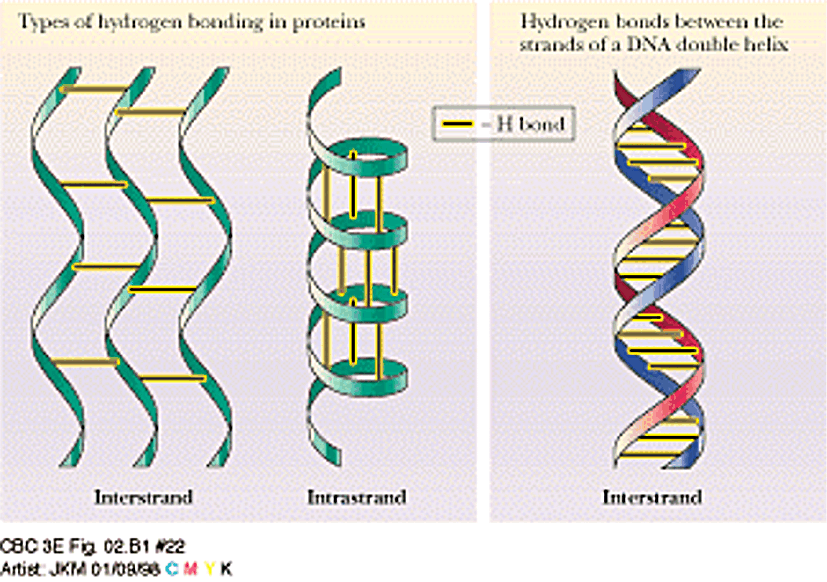
Secondary Structure:
local conformations of backbone, maintained by hydrogen bonds
Tertiary Structure:
3D structure of a subunit (one polypeptide chain) in its native
state
Quaternary Structure:
Spatial arrangement of subunits in oligomeric proteins
Denaturation:
Partial to complete unfolding of native conformation
Denatured Protein: Protein that has lost its native conformation
Charge-charge
Interactions (ion pairing, salt bridge) - strong force over long distance
Weakened by dielectric of water, stronger if buried in interior of
protein.
Polar
interactions - ~ charge - charge for molecules with permanent dipoles
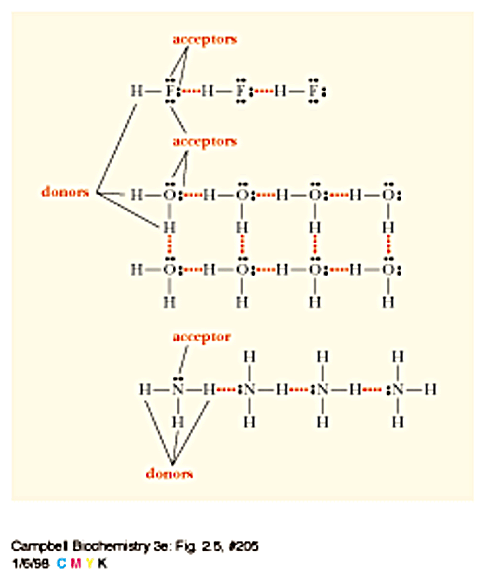
Hydrogen
bonds - moderate strength but directional (~5-20 kJ/mol)
Forces help stabilze structures of biomolecules - provide specificity
Hydrophobic
interactions - Weak but plentiful (~4-12 kJ/mol)
Van
der Waals Forces - permanent dipole/induced dipole; induced/induced dipoles
Weak but plentiful (~0.4 kJ/mol), very short range
Review:
Gibbs Free Energy
Gibbs
Free Energy
(DG
= DH
- TDS
)
"Spontaneous
processes are favored if they are energy "down hill" (i.e.
exothermic,
neg. DH
) and have an increase in entropy
(pos. DS)
(DG
< 0
- exergonic
/
"spontaneous")
(DG
= 0
- at equilibrium
(DG
> 0
- endergonic
/
"non-spontaneous")
DG
and Keq
(consider
A +
B < -- >
C + D)
Molecular Interactions and Protein Folding
Review of Thermo: DG = DH - TDS / van't Hoff plots
Thermo of "folding" - DS for unfolding is always (+); DH = ??
Unfolded polypeptide - at least 3 possible phi/psi angles per residue
S = RlnW = Rln(9)**99 = 1.8 kJ/mol-K for 100 a.a.
Native / folded protein - one phi/psi per residue
S = RlnW = Rln(1)**99 = 0 for native protein
DGconf = -TDSconf = 540 kJ/mol
DH term is dominated by many noncovalent interactions:
Review of Non-Covalent Interactions (see above)
Can Molecular Mechanics use Newtonian Physics to Predict Folded State?
Need to define Molecular Force Fields - find Emin using Molecular Dynamics
Define Bonding and Non-bonding Potentials
Non-bonding Potentials:
Electrostatic - role of "D", the dielectric
Dipole-dipole interactions - important in alpha helices (aligned dipoles)
vander Waals (dispersion forces) - weak but numerous
H-bonds - give specificity to interactions
Hydrophobic Interactions / Conformational Entropy
We can use the various potentials to find preferred conformational angles, dipole interactions, etc., but since this is a many variable problem, and solute-solute interactions are often balanced by solute-solvent interactions, this has not been very successful to date in predicting protein structure. However, these potentials are very useful to optimize a structure once a fairly good starting model is found (X-ray, NMR, etc.). To improve the "convergence" of such methods, molecular simulations are used to allow the molecule to sample greater conformational space. In the computer, the molecule can be "heated" to very high temperatures (3000o) to give it sufficient energy to overcome local minima and sample alternative conformational spaces. It is allowed to sample this larger conformational space for several nanoseconds (torsion angles twisting occurs in psec) in the computer simulation and then slowly "annealed" by lowering the temperature in steps while doing energy minimizations.
Protein
Folding: Stability / Denaturation and Renaturation
Protein
folding (tertiary structure) is determined by weak interactions
DG = Gf
- Gu
= DHprot
+
DHsolv
-TDSprot - TDSsolv
( largest - TDSsolv
for nonpolar R)
Folding
as a cooperative, sequential process : Local sec. st. / Domains / Molten
globules
Molecular
chaperones : (GroEL , GroES) assist with folding of some proteins
Renaturation
: Chris Anfinsen - Folding of Ribonuclease (4 disulfides)
Folding Pathway -
Levinthal Hypothesis - How can proteins fold in a finite amount of time if they have to sample large regions of conformational space?
New Model - use of "molten globules" of pronounced, local secondary structure but of loose tertiary structure. As folding proceeds, steps lead to more compact tertiary structures that allow the expulsion of solvent and the tight packing of hydrophobic groups in the interior away from solvent. Molecular Chaperones help enable the folding process.
Predicting Protein Folding
1. Molecular - Dynamics Simulations
2. Monte-Carlo Simulations
3. Statistical Methods - use database of known structures
Chou / Fasman - 1974 - Propensity for a.a. residues to be in helix, sheet, or turns.
Many web-based programs that allow one to input an amino acid sequence and look at the predicted secondary structure averaged over many computational procedures.
Use of Molecular Transitions to Study the Energetics of Folding
Many biomolecules undergo sharp transitions (proteins: coil / alpha helix ; DNA: d.s. vs. s.s.). Often these structural transitions can be followed using CD spectroscopy. Such sharp transitions are usually the result of cooperative (all-or-none) transitions. The "zipper" model attempts to model this process by dividing the rates into a slow "nucleation" step followed by rapid "propagation" steps. Following these transitions as a function of Temperature allows one to plot logK vs. 1/T to obtain thermodynamic information (-DH/R) about folding from a van't Hoff plot.
Denaturation (Non-native state): There are many denatured states of macromolecules. Denaturation can occur from many causes:
Denaturation : heat, high salt, hi & lo pH, organic solv., mechanic
- Tm (melting temperature)
- 8M Urea ; 5M quanidinium chloride ; 1% SDS
- Anions : sulfate >
phosphate > Cl- > Br- > SCN-
- Cations: ammonium >
Cs+ > K+ > Na+ > Li+ > Mg2+ > Ca2+ > Ba2+
{kind=link}
{kind=link}