Mass spectrometry: an
introduction
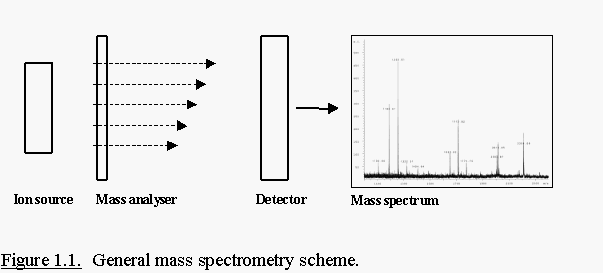
Mass spectrometers provide the ability to accurately measure the mass
of almost any molecule that can be ionised to the gas phase. A mass
spectrometer consists of three essential components (Fig. 1.1.); an ion
source, a mass analyser and a detector. An ion source converts molecules
into gas-phase ions. Once these ions are created, they are separated in
the mass analyser by their mass (m) to charge (z) ratio and detected by an
electron multiplier. MS data are recorded as ‘spectra’ which display ion
intensities versus their m/z value.
MALDI-TOF-MS: technology
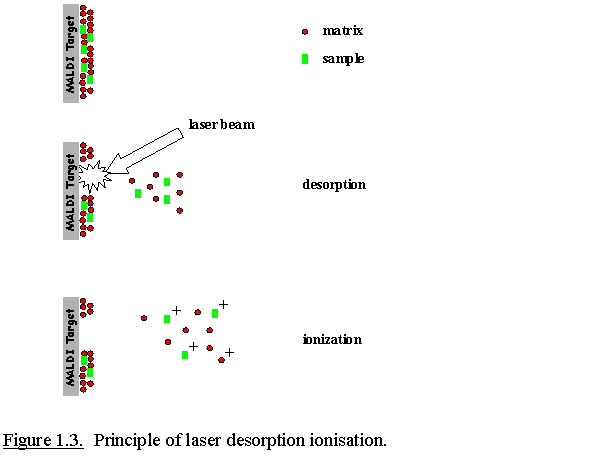
Samples that are analysed
by MALDI (matrix-assisted laser desorption ionisation) are first mixed
with so called matrix molecules and spotted on an AnchorChip target or a
stainless steel target. Upon drying, the matrix molecules crystallise and
solid sample/matrix co-crystals are finally formed (Fig. 1.2.). The
MALDI-target is then inserted into the ion source of the mass spectrometer
which is under a high vacuum. A strong electrical field is applied between
the target and the extraction plate(s). A laser (e.g., a pulsed nitrogen
laser at 337 nm) is fired onto the sample, resulting in a desorption event
due to absorbance of the laser energy by the matrix molecules. Energy
deposition into the matrix molecules results in the modulation of the
absorbed energy into heat. This rapid heating causes sublimation of the
matrix crystals and subsequent expansion of matrix molecules and the
co-crystallised analyte molecules into the gas phase. The ions are
repelled from the target surface and accelerated into the mass analyser
(Fig. 1.3.).

In the MALDI ionisation process singly-charged ions are mainly formed
by protonation of the basic residues such as the side chains of arginine,
lysine, histidine and the free alfa-amino group. MALDI is moderately
tolerant towards contaminants such as salts and detergents. Nevertheless
it is still advisable to remove as many contaminants from a sample as
possible since they might compete with the peptides/proteins for
ionisation and might interfere with the crystallisation process.
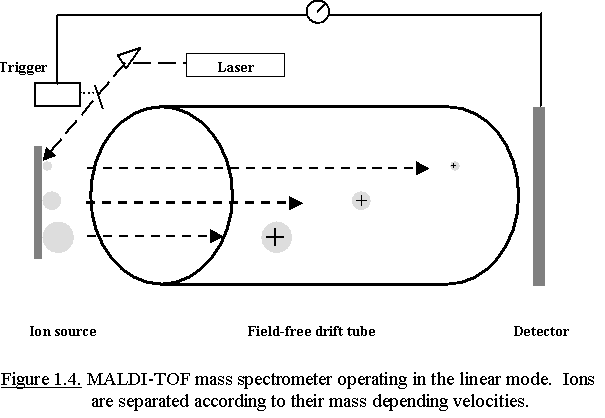
Usually, MALDI is coupled to a time-of-flight (TOF) tube for mass
analysis. The TOF tube is under a high vacuum (10-6 - 10-8 mbar) and is a
field-free drift region. All ions entering the TOF tube have a fixed
kinetic energy; => Ekin = ½ m.v² = q.V = constant where V =
acceleration potential and q = the charge of the ion (=z)
The ions pass the field-free drift tube with a velocity proportional to
(m/z)-1/2. This implicates the higher the mass of the ion, the lower its
velocity and the longer it takes before the ion strikes to the detector.
Based upon their different velocities ions of different mass can be
separated during their flight in the TOF-tube. A detector that has been
triggered by the laser pulse records the time-of-flight of the ions (Fig.
1.4.). Smaller ions fly faster than larger ions, and their m/z ratio can
be calculated from their flight time after calibration of the analyser
using compounds with known mass.
Inherent to the MALDI ionization process is a spread of kinetic energy
of ions; resulting in different points in time and space of ion formation
within the ion source. Thereby ions with the same mass obtain different
kinetic energies and velocities during their extraction out of the ion
source. This results in peak broadening of the ion signal at the detector.
Thereby the mass resolution, which is a measure of an instruments
capability to produce separate signals (isotopic peaks) from ions of
similar mass, is limited.
This peak broadening can be reduced by the use of an ion mirror (a
reflectron) at the end of the linear flight tube and by delayed ion
extraction out of the ion source.
With delayed extraction (DE), the
extraction voltage pulse is applied between 100 and 500 ns following the
laser pulse. During this delay, ions are allowed to spread in the source
and higher energy ions will move further away than lower energy ions with
the same mass. The extraction voltage is now applied as a potential
gradient over the ion source. This compensates for the distribution of
initial kinetic energies, so that ions with identical m/z values will
arrive at the same time at the detector.
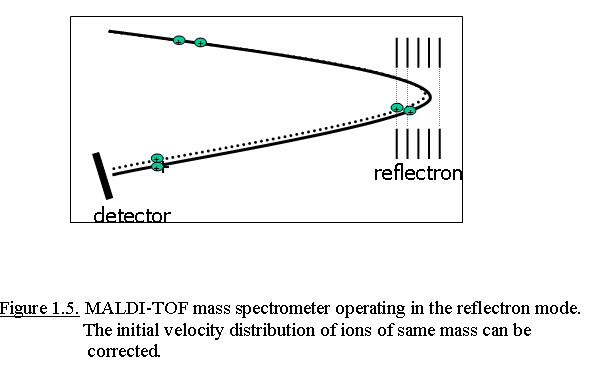
A schematic representation of an ion mirror is shown in Fig. 1.5. The
reflectron has an applied voltage higher than that of the accelerating
voltage in the ion source, resulting in a slowing down of incoming ions,
and finally a reversion of their flight path to the second detector. Ions
with a slightly lower kinetic energy do not penetrate the reflectron as
deep and thus turn around faster, catching up with ions of slightly
greater kinetic energy that penetrate the reflectron deeper. Thereby the
flight times of ions with an identical m/z values, but different Ekin
values will be corrected when the ions strike to the detector.
MALDI-TOF-MS: applications
A MALDI-TOF instrument
can produce two different types of data that can be used for protein
identification/characterization. The first type of information is the
accurate measurement of the molecular weight of peptides: Peptide Mass
Fingerprinting (PMF). Sequence information on peptides can be generated by
Post-Source Decay (PSD) analysis of selected peptides.
- Peptide Mass Fingerprint analysis
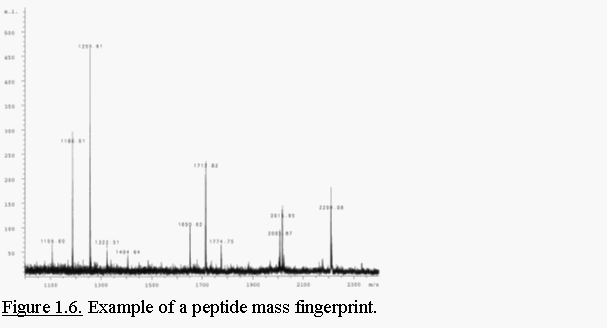
Digestion of a protein
with a protease, produces a set of peptides that serves as an unique
fingerprint of the given protein (Fig. 1.6.). In MALDI-TOF-MS peptides
generally carry one charge and consequently show only one peak in a m/z
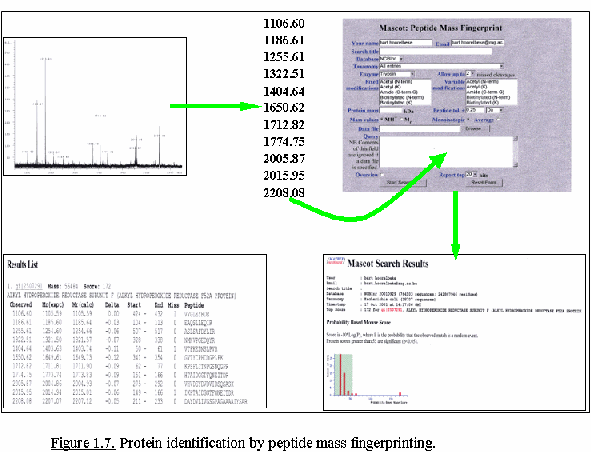
spectrum which facilitates data interpretation. Protein identification
by PMF (Fig. 1.7.) is widely used for peptide mixtures produced by
in-gel tryptic digests. PMF is the method-of-choice for protein
identification in proteome studies because it is a simple and sensitive
technique (femtomole amounts), and is gradually reaching a confidence
level comparable to sequence-based identification. This is due to
improved mass accuracy and availability of complete genomic information
for a growing number of model organisms.
- Post-Source Decay analysis
A second method employs
peptide fragmentation data that is generated by PSD analysis. In this
method, data, specific for an individual selected peptide is collected.
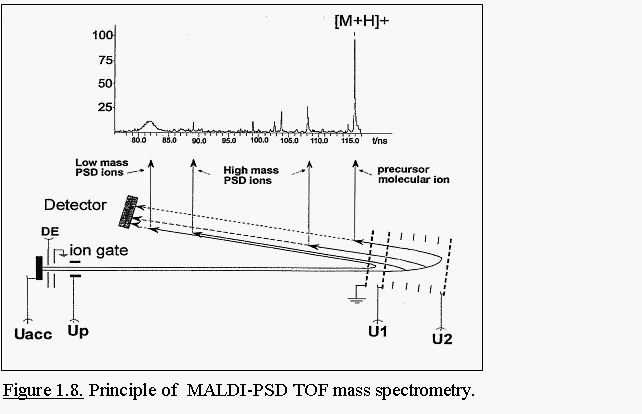
The principle setup of a mass spectrometer for PSD analysis is
illustrated in Fig. 1.8. Ions leaving the ion source and having acquired
sufficient internal energy during the desorption process, can release
this energy by undergoing a certain degree of decomposition into product
ions during their flight in the TOF tube. Since the decomposition of the
precursor ion happens in a field-free region, the daughter ions still
have the same velocity as their precursor ion. In linear MALDI
instruments, PSD ions are therefore detected at the same time as their
parent ions. Due to their lower mass, PSD ions have a much lower kinetic
energy than their precursors. The ion reflector is in a PSD analysis
used as an energy filter and thus a mass analyser for PSD ions.
Most
reflectron fields have a limited dynamic range; this means that not all
PSD-generated fragment ions will hit the detector. A complete PSD
spectrum is therefore acquired in several steps of reflector potentials.
Finally all sections of the PSD spectrum are stitched
together.
Precursor ion selection is realised by ‘ion gating’. All
ions passing the ion gate are electrostatically deflected except ions in
a well-defined mass window that is transmitted without deflection by
turning off the deflection pulse. Peptides can often physically selected
from the same sample used to generate a peptide mass fingerprint; so
there is no additional peptide or protein material required.
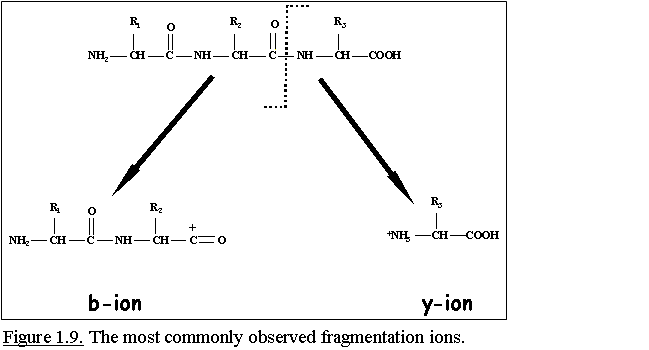
MS fragmentation of peptides primarily occurs at the amide
bond (-CO-NH-) between two amino acid residues. The most commonly
observed fragmentation ions are b and y ions. B ions are
sequence-specific fragment ions derived from the N-terminus and y ions
are from the C-terminus (Fig. 1.9.).
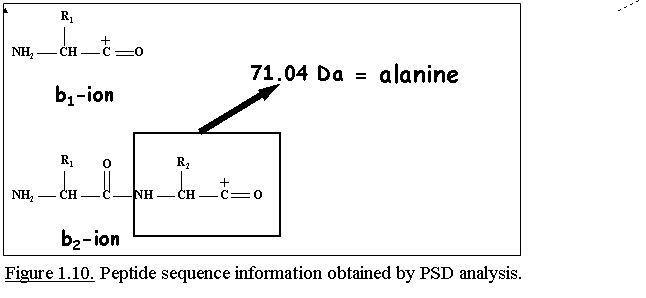
Since all 20 naturally occurring amino acids have different
masses, except Ile/Leu and Lys/Gln, the observed mass difference between
two consecutive fragment ions of the same type determines the identity
of the amino acid because this mass difference depends of the nature of
the side chain of the amino acid (Fig. 1.10.).
PSD spectra are composed of b and y ions, internal fragment
ions, immonium ions (= low mass fragment ions from single amino acids)
and the precursor ion. Since MALDI-PSD is an unimolecular decay, the
efficiency of peptide fragmentation is very low. This results in a
rather low quality of the spectra containing a complex fragmentation
pattern and a high background noise level (Fig. 1.11.).
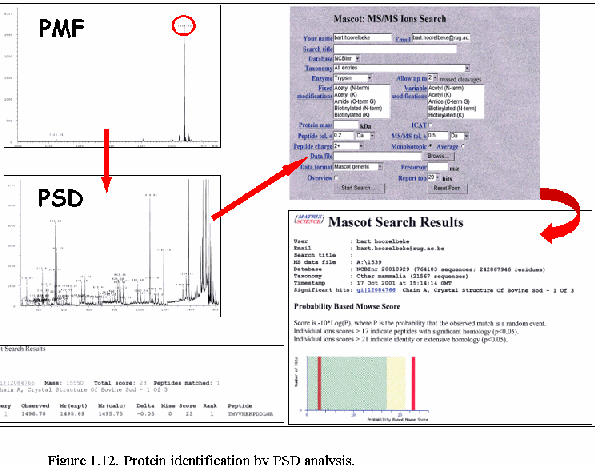
Knowing the mass of the peptide, some fragmentation data
(sequence data), and the cleavage specificity of the protease employed
has proven to be sufficient to identify peptides by database searching
(Fig. 1.12.). One or more PSD spectra matching to sequences in the same
protein, provide a high level of confidence in the identification.