Protein Expression, Purification and Analysis
To
study proteins and their functions, one must first Produce,
Extract, and Purify the protein.
Produce - tissue
rich in protein / over-expression using cultured cells (see below)
Extract - cell
disruption followed by centrifugation
Purification -
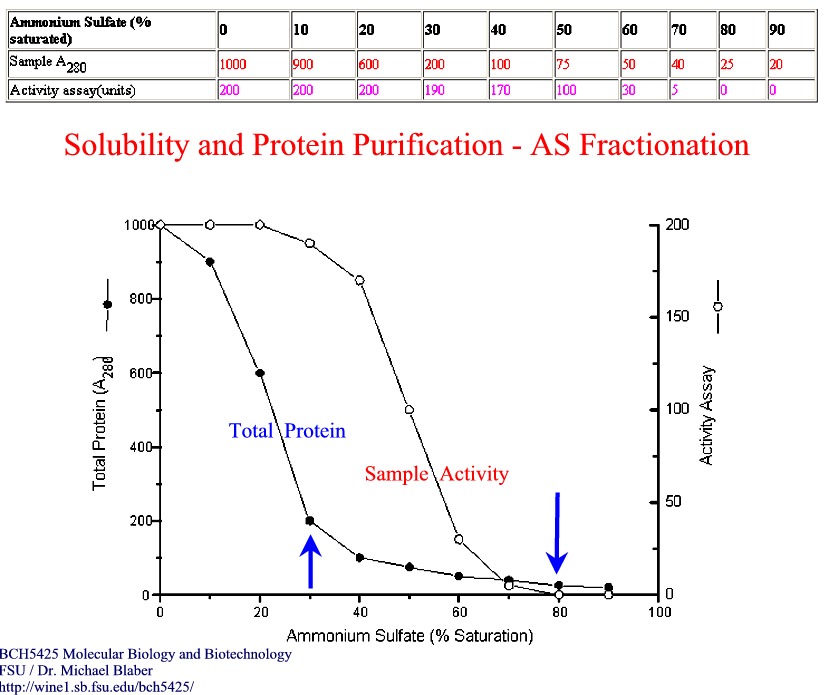
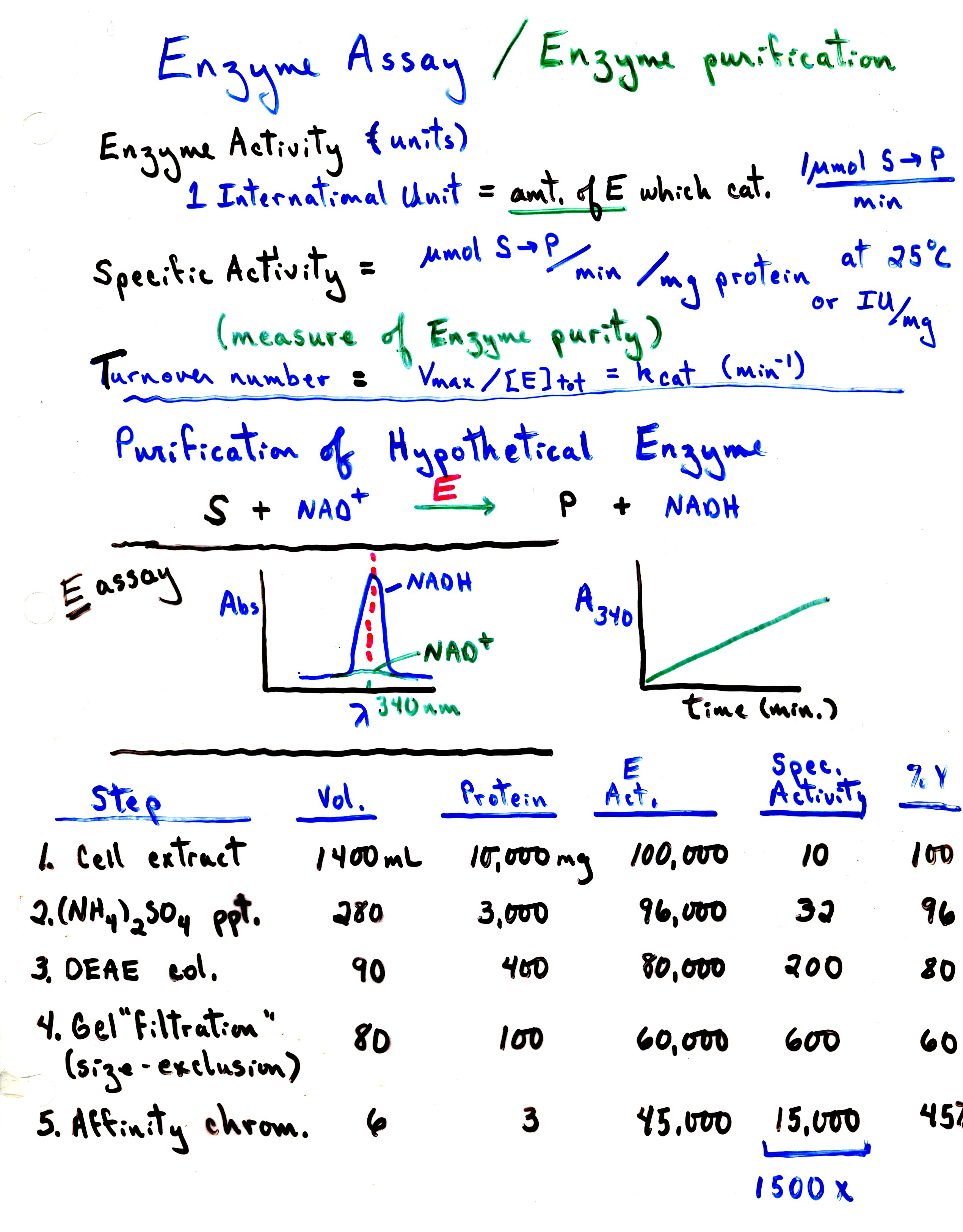
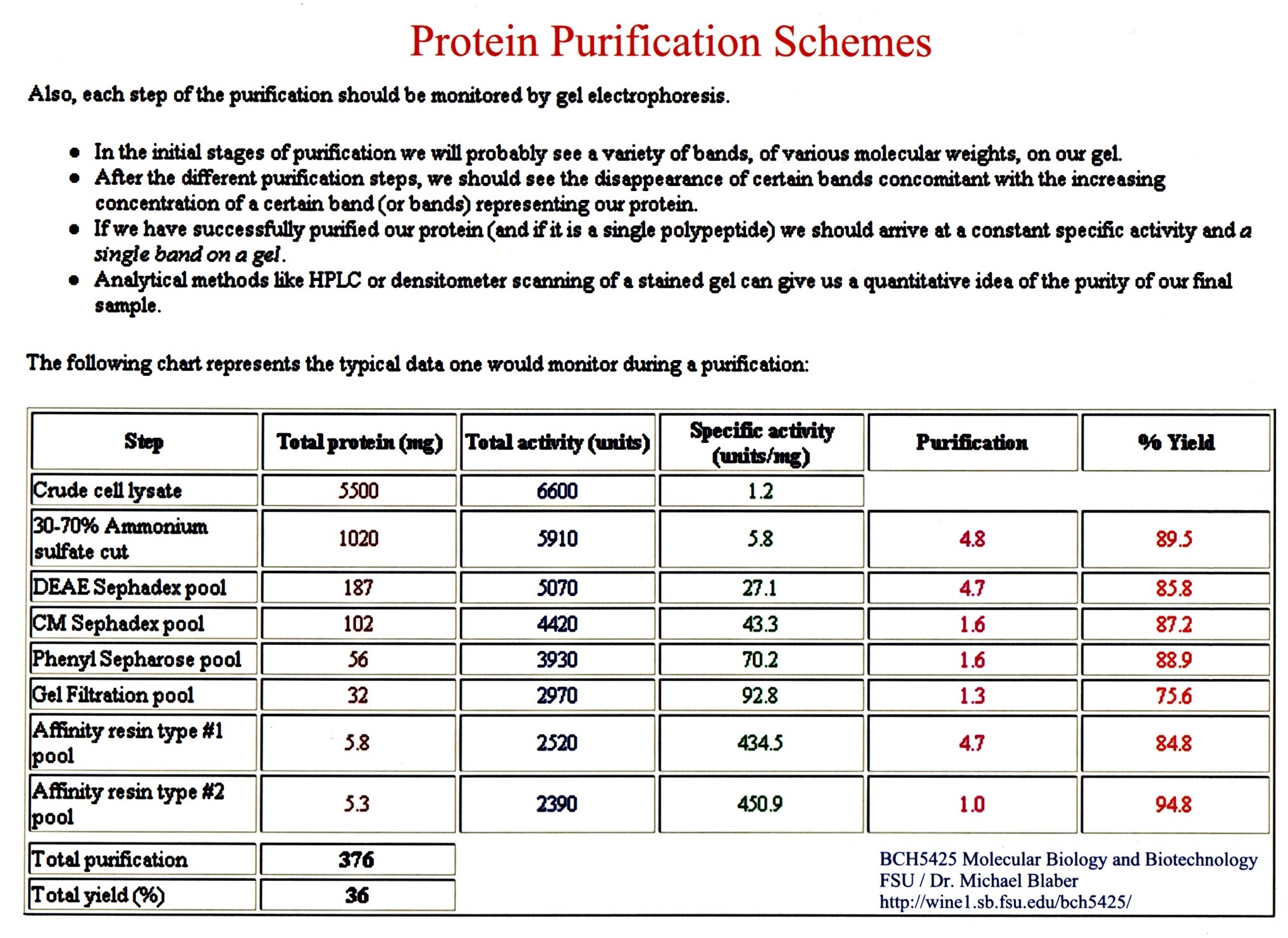
take advantages of differences in solubility, charge, size, and specificity.
(An assay
is needed to monitor the progress of the purification process.)
********************************
In
olden days, protein purification used to start with a source (organ
tissue, plant source, bacteria line) known to be rich in the protein of
interest. Often this would involve
obtaining many pounds of organ tissue directly from a slaughter house or growing
large (30 or 60L) batches of bacterial culture in order to isolate sufficient
material for further studies. Purification
based on solubility and charge.
Typical protocol for isolation of a mammalian protein (after
procedure for chicken heart LDH H4, Nathan
Kaplan et al., JBC, 239: 1753-1761 (1964):
Step 1: Obtain
25 lbs of fresh chicken hearts on ice from local slaughter-house.
Step 2: Cut
tissue in small sections, grind tissue in a commercial meat grinder, extract
with 14L cold distilled water.
Step 3: Filter
cell and connective tissue debris from extract by passing thru double layer of
cheese cloth.
Step 4: Centrifuge
using 1L containers and large bucket rotor International centrifuge at 1300 x g
for 30 minutes. Pool supernatant.
Assay.
Step 5: Adjust
pH to 7.0, add solid ammonium sulfate to give 70% saturation of AS, maintain pH
~ 7 by adding dilute ammonium hydroxide as needed. Let stand for 2-4 hours, filter overnight on fluted filter
paper. Scrape ppt. off filter paper
and suspend in 1 L of cold distilled water.
Centrifuge. Assay.
Step 6: Dialyze
soluble fraction against two 20 L changes of 0.005M Tris buffer, pH 7.0 for a
total of 12 hours. To the solution,
add solid AS to 25% saturation. Centrifuge,
remove ppt. Add more AS to 60%
saturation, leave standing for 1 hour, spin at 18,000 x g for in a Sorvall
refrig. Centrifuge, save pellet. Dissolve
pellet in 300 mL of 0.005 M Tris buffer, pH 7.6.
Dialyze against two 20 L changes of 5 mM Tris, pH 7.0 for 8 hours each,
centrifuge.
Step 7: Add
precooled (-15 deg) acetone slowly to dialysate to ppt protein at 25%/(v/v).
After 10 minutes, cent. discard ppt.
Add more acetone to 50% (v/v), leave for 20 minutes at 0oC.
Collect ppt. At 18,000 x g for 30 min., then extract with 200 mL cold
Tris buffer, pH 7.6, cent., discard ppt.
Step 7: Add
supernatant to DEAE cellulose column. Elute
with a gradient of 2 L each of 0.005 M Tris, pH 7.0 and same buffer with 0.20 M
NaCl. Combine fractions with LDH
activity. Assay.
Expected yield 50-60 %.
Step 8: Re-crystallization
- Ppt. with saturated AS, redissolve in a minimum of buffer, centrifuge, add AS
slowly with stirring to 30% saturation. Wait
about 1 day, centrifuge to harvest enzyme.
Assay. Yield ~ 30%, 100X
purification.
***********************
Today, the majority of proteins being studied in the laboratory
take advantage of more modern tools of biotechnology to produce
large quantities of proteins needed for study.
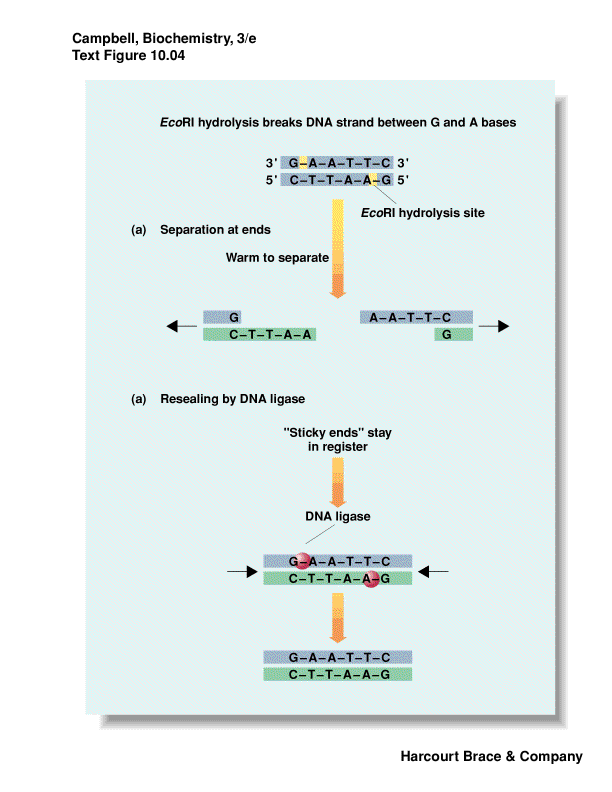
Restriction enzyme--molecular scissors
-
endonucleases--does not require an end (exonucleases)
-
>100 restriction enzymes known
-
names come from organism:
-
recognize a specific palindromic DNA sequence and cut the DNA
-
palindrome is the same forwards/backwards
-
some leave 3' overhang; 5' overhang or blunt ends
-
overhangs leave--"sticky ends"--even though DNA is cut, can have base-pairing
-
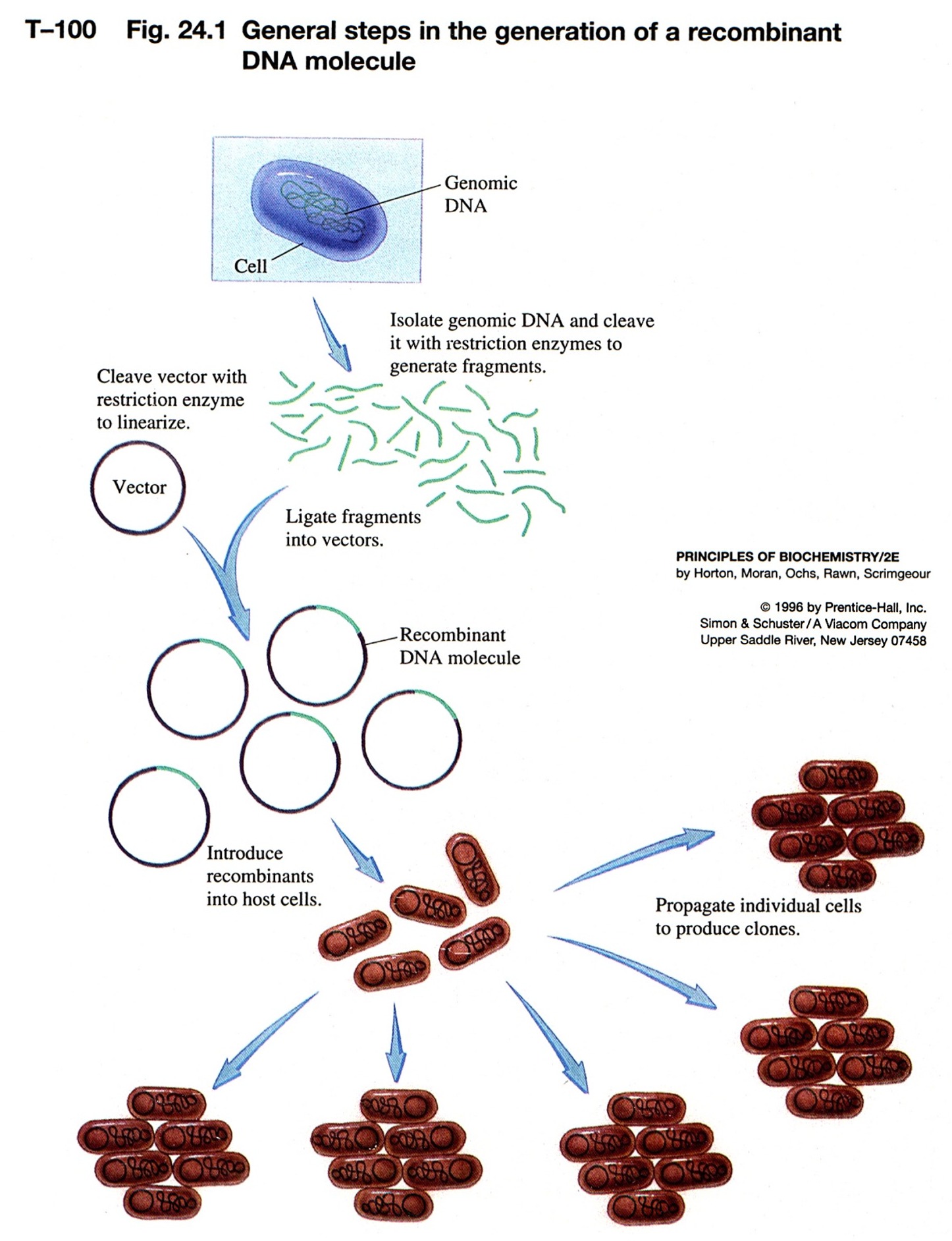
move DNA from one organism to another - "recombinant DNA"
-
put DNA together with DNA ligase
-
use synthetic DNA of desired sequence to "paste" on restriction site if
nature did not provide one
|
-
methylation protects DNA from restriction enzymes
-
mechanism for bacteria to protect itself from invading phage or other bacterial
DNA
 |
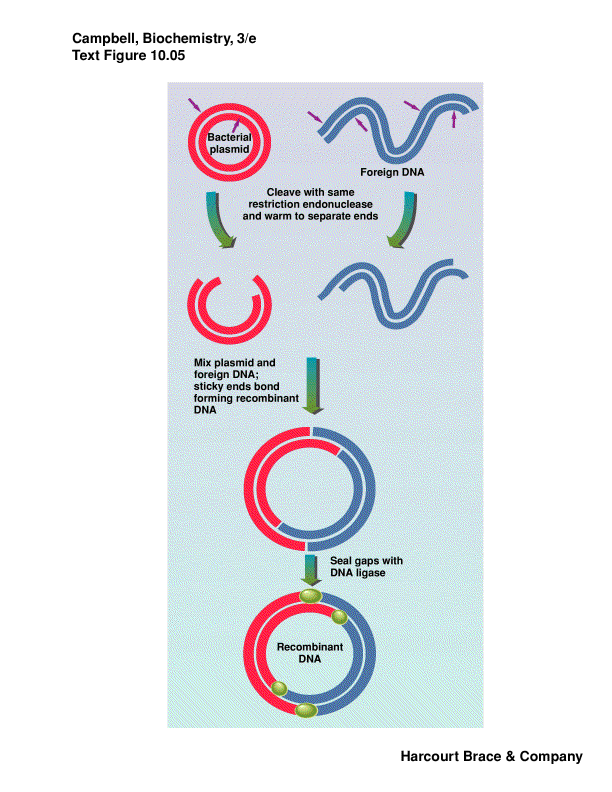
Plamids are cloning vectors
-
plasmids are closed circular DNA, with origin of replication--replicated
within bacteria to many copies
-
carries a resistance gene--ampicillin, tetracyclin, kanamycin
-
take DNA from one organism, cut with RE, isolate fragment desired from
a gel
-
cut a plasmid or phage DNA with same RE
-
put these two DNA fragments together via sticky ends, ligate them closed
-
we have recombinant DNA
-
this is transferred into bacterial cells by electroporation or chemical
competence
-
plate on media with antibiotic to kill bacteria that did not take up a
plasmid--no proof that your foreign DNA is there, only that the plasmid
is there
-
individual colonies contain a single plasmid
|
 |
-
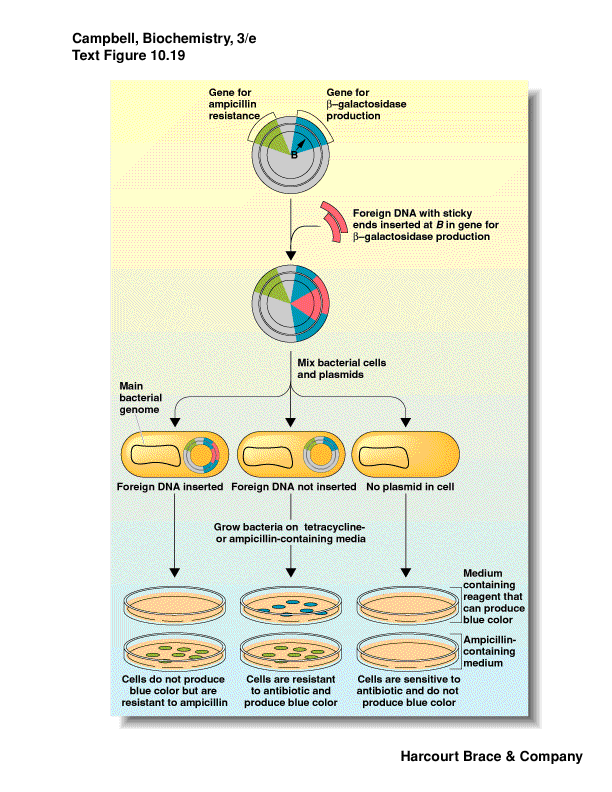
How do you know your foreign DNA was inserted?
-
one method: interrupt a gene that is a reporter - b-galactosidase (lacZ)
-
use a substrate for b-galactosidase that when
cleaved give a colored compound
-
do this on antibiotic media to select for plasmid
-
induce the gene with a lactose-analog
-
if the gene is intact get blue color--no foreign insert, just plasmid
-
if the gene has an insert (foreign DNA) then the reading frame is thrown
off and no b-galactosidase is produced--no color
|
 |
Purification -
take advantages of differences in solubility,
charge, size, and
specificity.
1. Solubility
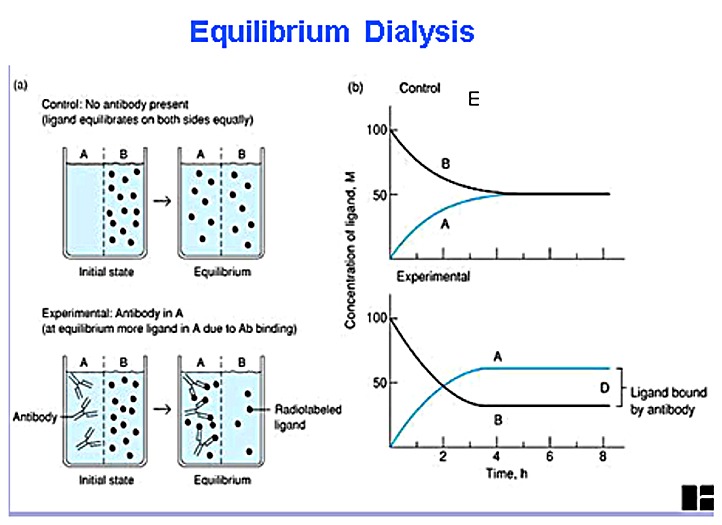
2. Size - Dialysis (figure)
3.
Column
chromatography
- Separation by charge / size / or
affinity
-
a matrix is in a cylindrical holder (Figure)
-
buffer flows through the matrix
-
fractions are collected
-
separation of biomolecules
Ion-exchange chromatography
-
proteins have charges due to amino acid side groups
-
bind to charged column matrix depending on their charge at a particular
pH
-
anionic--negatively charged: phosphocellulose, heparin sepharose, S-sepharose
-
cationic--positively charged: DEAE-sepharose, Q-sepharose
-
elute bound proteins from column based on charge and displacement by salt
or pH
High Performance Liquid Chromatography (HPLC)
-
gravity flow very slow--depends on size and amount of liquid at the top
-
HPLC used high pressure to force liquid through
-
special matrixes and columns
-
fast and sometimes better resolution
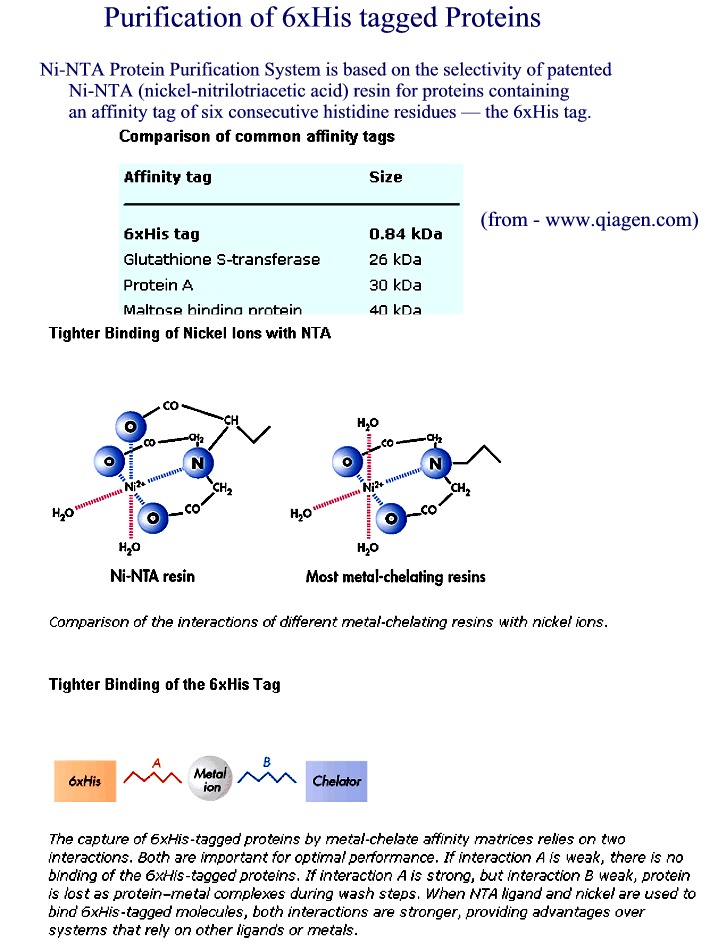
Affinity Chromatography - use of "tagged"
proteins to create affinity site - sep. by specificity
-
column matrix has a ligand that specifically binds a protein
-
specialty affinity columns for binding recombinant proteins with certain
"tags"
-
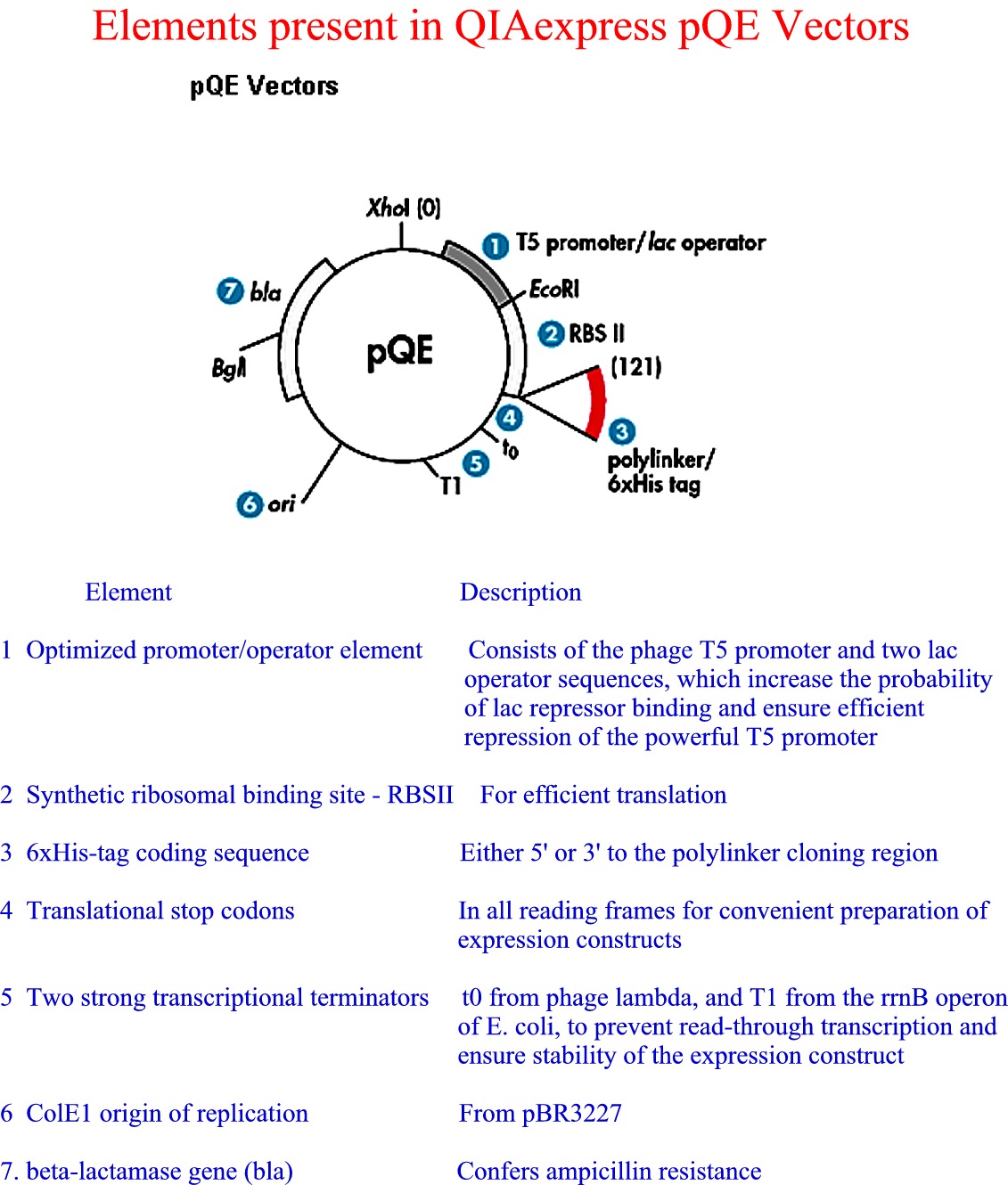
6xHis added at N or C terminus--binds Ni++ column
-
His tag (Figure 1) (Figure
2) (Figure 3) (Figure 4)
-
other types of "tags"--chitin, glutathione S-transferase (GST).....
|
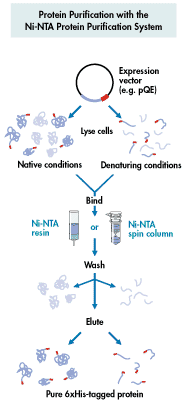
from Qiagen
website |
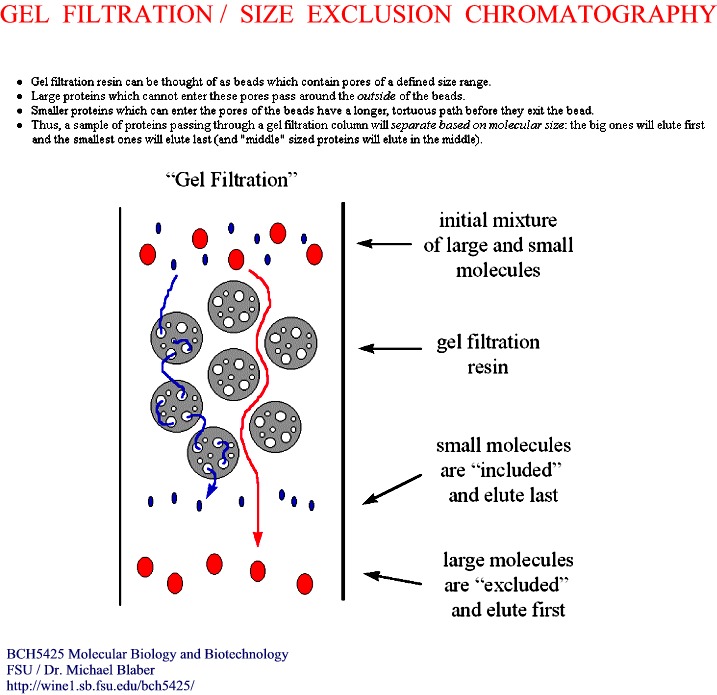
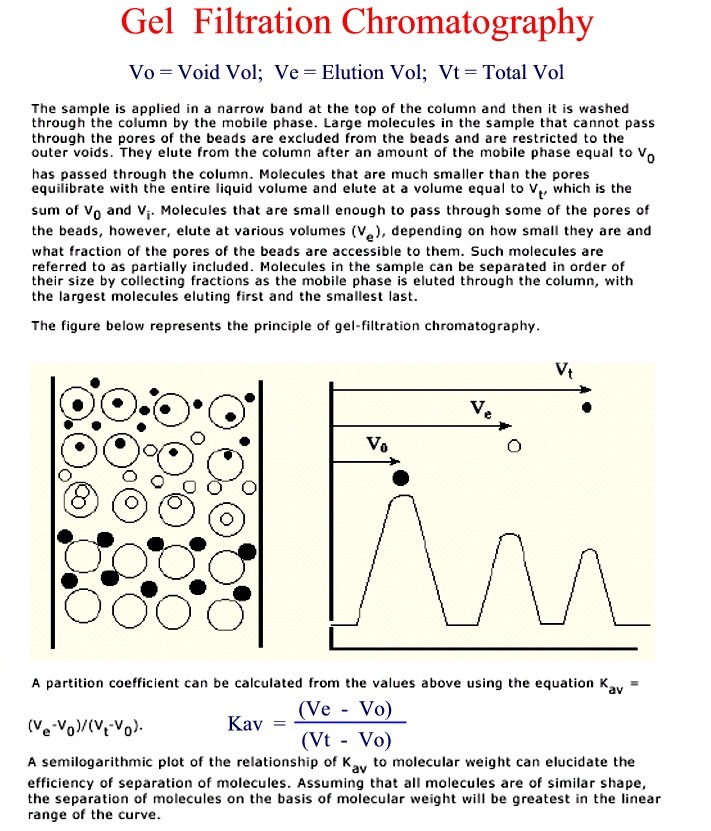
-
separates on the basis of size, not charge
-
porous beads--think of golf balls
-
small molecules go into the holes and get trapped temporarily (Figure)
-
large molecules are too large to enter the holes and pass on by
-
exclusion size--depends on the size of the holes
-
how long the molecules get trapped determines elution order
-
large out first > medium > small out last (Figure)
-
choose the size of matrix for the separation needed
-
Terms: Vtot, Vo, Vpoly, Ve, Kav = (Ve - Vo)/(Vt - Vo)
4. Electrophoresis
(theory and more detail on this later)
-
method of separation of charged molecules
-
movement of charge particles in an electrical field
-
electrophoretic mobility reflects both charge and size/shape
-
Many kinds of electrophoresis: usually have a solid medium e.g.,
paper, gel, TLC plate
-
gel electrophoresis used to separate large molecules: DNA, RNA, protein
-
a "gel" is like very stiff JELLO
-
acrylamide forms polymers that can be cross-linked to varying degrees in
a chemical process
-
the cross-linking determines the size of "holes" that the molecules pass
through
-
apply electrical current across the medium, Anode (+) and Cathode (-)
-
positively charged molecules move to the Cathode
-
negatively charged molecules move to the Anode
-
proteins depending on their charge will move in either direction,
SDS (sodium dodecyl
sulfate) gels (more detail on this later)
-
SDS is a detergent--amphipathic, has both hydrophobic and hydrophilic characteristics
-
hydrophobic tail of SDS interacts with hydrophobic side chains of amino
acids
-
number of SDS molecules bound is proportional to the number of amino acids
-
1 molecule of SDS bound per ~2 amino acids
-
SDS overwhelms any inherent charges and effectively turns the protein into
a polyelectrolyte
-
SDS also disrupts tertiary structure
-
b-mercaptoethanol is also included to reduce
disulfide bonds and destroy intra-chain cross-linking
-
protein mobility in SDS PAGE is proportional to protein size
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}