Tools - Electrophoresis and Chromatography
To
study proteins and their functions, one must first Produce,
Extract, and Purify the protein.
Produce - tissue
rich in protein / over-expression using cultured cells (see below)
Extract - cell
disruption followed by centrifugation
Purification -
take advantages of differences in solubility, charge, size, and specificity.
********************************
Column
chromatography
- Separation by charge / size / or affinity
-
a matrix is in a cylindrical holder
-
buffer flows through the matrix
-
fractions are collected
-
separation of biomolecules
Ion-exchange chromatography
-
proteins have charges due to amino acid side groups
-
bind to charged column matrix depending on their charge at a particular
pH
-
anionic--negatively charged: phosphocellulose, heparin sepharose, S-sepharose
-
cationic--positively charged: DEAE-sepharose, Q-sepharose
-
elute bound proteins from column based on charge and displacement by salt
or pH
High Performance Liquid Chromatography (HPLC)
-
gravity flow very slow--depends on size and amount of liquid at the top
-
HPLC used high pressure to force liquid through
-
special matrixes and columns
-
fast and sometimes better resolution
Affinity Chromatography - use of "tagged"
proteins to create affinity site - sep. by specificity
-
column matrix has a ligand that specifically binds a protein
-
specialty affinity columns for binding recombinant proteins with certain
"tags"
-
6xHis added at N or C terminus--binds Ni++ column
-
other types of "tags"--chitin, glutathione S-transferase (GST).....
|
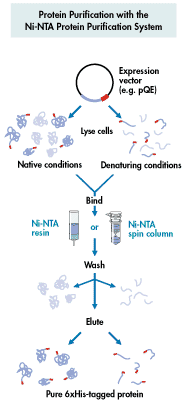
from Qiagen
website |
-
separates on the basis of size, not charge
-
porous beads--think of golf balls
-
small molecules go into the holes and get trapped temporarily
-
large molecules are too large to enter the holes and pass on by
-
exclusion size--depends on the size of the holes
-
how long the molecules get trapped determines elution order
-
large out first > medium > small out last
-
choose the size of matrix for the separation needed
There are many biochemical / biotech suppliers with excellent web sites with
product information and tutorials on these techniques - such as this link to
Amersham Biosciences web page on chromatography.
Electrophoresis
-
method of separation of charged molecules
-
movement of charge particles in an electrical field
-
electrophoretic mobility reflects both charge and size/shape
-
Many kinds of electrophoresis: usually have a solid medium e.g.,
paper, gel, TLC plate
-
gel electrophoresis used to separate large molecules: DNA, RNA, protein
-
a "gel" is like very stiff JELLO
-
acrylamide forms polymers that can be cross-linked to varying degrees in
a chemical process
-
the cross-linking determines the size of "holes" that the molecules pass
through
-
apply electrical current across the medium, Anode (+) and Cathode (-)
-
positively charged molecules move to the Cathode
-
negatively charged molecules move to the Anode
-
proteins depending on their charge will move in either direction,
SDS (sodium dodecyl
sulfate) gels
-
SDS is a detergent--amphipathic, has both hydrophobic and hydrophilic characteristics
-
hydrophobic tail of SDS interacts with hydrophobic side chains of amino
acids
-
number of SDS molecules bound is proportional to the number of amino acids
-
1 molecule of SDS bound per ~2 amino acids
-
SDS overwhelms any inherent charges and effectively turns the protein into
a polyelectrolyte
-
SDS also disrupts tertiary structure
-
b-mercaptoethanol is also included to reduce
disulfide bonds and destroy intra-chain cross-linking
-
protein mobility in SDS PAGE is proportional to protein size
The following is after the Amersham
Biosciences Web Site: In SDS polyacrylamide gel
electrophoresis (SDS-PAGE) separations, migration is determined not
by intrinsic electric charge of polypeptides but by molecular
weight. Sodium dodecylsulphate (SDS) is an
anionic detergent that denatures proteins by wrapping the hydrophobic
tail around the polypeptide backbone. For almost all proteins, SDS binds at a
ratio of approximately 1.4 g SDS per gram of protein,
thus conferring a net negative charge to the polypeptide in proportion to its
length. The SDS also disrupts hydrogen bonds, blocks hydrophobic interactions,
and substantially unfolds the protein molecules, minimizing differences in
molecular form by eliminating the tertiary and secondary structures. The
proteins can be totally unfolded when a reducing agent such as dithiothreitol (DTT)
is employed. DTT cleaves any disulphide bonds between cysteine residues. The SDS-denatured
and reduced polypeptides are flexible rods with uniform negative charge per unit
length. Thus, because molecular weight is essentially a linear function of
peptide chain length, in sieving gels the proteins separate by molecular weight.
There are two types of buffer systems used in protein gel electrophoresis: continuous
and discontinuous.
A continuous system uses only one buffer for
the tanks and the gel. In a discontinuous system, first developed by Ornstein
(1964) and Davis (1964), a nonrestrictive large-pore gel called a stacking gel
is layered on top of a separating (resolving) gel. The two gel layers are each
made with a different buffer, and the tank buffers differ from the gel buffers.
In a discontinuous
system, protein mobility a quantitative measure of the migration
rate of a charged species in an electric field is
intermediate between the mobility of the buffer ion of the same charge (usually
negative) in the stacking gel (leading ion) and the mobility of the buffer ion
in the upper tank (trailing ion). When electrophoresis is started,
the ions and the proteins begin migrating into the stacking gel. The proteins
concentrate in a very thin zone, called the stack, between the leading ion and
the trailing ion. The proteins continue to migrate in the stack until they reach
the separating gel. At that point, due to a pH or an ion change, the proteins
become the trailing ion and "unstack" as they separate on the gel.
Although a continuous system is slightly easier to set up than a discontinuous
system and tends to have fewer sample precipitation and aggregation problems,
much greater resolution can be obtained with a discontinuous system. Only
minimal concentration of the sample takes place with continuous gels, and
proteins form zones nearly as broad as the height of the original samples in the
sample wells, resulting in much lower resolution. The discontinuous
Laemmli system (Laemmli, 1970), a denaturing modification of Ornstein (1964) and
Davis (1964), is the most widely used system for
research protein electrophoresis today. The resolution in a Laemmli
gel is excellent because the treated peptides are concentrated in a stacking
zone before entering the separating gel.
| Caution: |
Acrylamide
is a neurotoxin and should be handled with care. |